2014-04-14 识林
关于药品招标、医保报销问题一直讨论不断,在医保报销中,监管部门又可以提供怎样的参考?翻出去年的一篇旧闻,研究对象虽是研究分子诊断产品,但相信现在读来仍有些许启发。期望能有一个办法彻底解决问题是太过乐观了,但一些“可能”与“希望”总是有的。正如文中所说“分子诊断所面临的监管和医保政策困境的最终解决方案,也许需要法律或规章的彻底改变,而这一改变在当前的政治经济环境下不可能发生。与此同时,在当前的法律和规章框架下,渐进而具有价值的进展却是可能的。尤其是两个创新性的政策措施——CED(“依据证据发展的医保覆盖”)和平行审评——为FDA和CMS(医疗保险和补助服务中心,Centers for Medicare & Medicaid Services)减少创新医疗产品的评估过程中的关键瓶颈带来了希望。”
监管与报销创新
针对分子诊断辅助设备监管和医保报销的“依据证据发展的医保覆盖”政策与“平行审评”政策
2013年3月13日发表于美国Science的 www.sciencetranslationmedicine.org ,英文原文![]()
分子诊断检测在医疗保健向着更加有效、更具效率和个性化发展的进程中扮演着日益关键的角色。这些检测将通过使卫生保健提供商针对个体患者优化疗法来改善民众健康状况,受到广泛期待。然而,尽管分子诊断检测能够改善医疗保健,但却面临来自监管和医保报销政策的挑战,令其发展受阻,也妨碍与常规医疗保健服务体系的融合。
两个创新方案:“依据证据发展的医保覆盖”(CED)和“平行审评”,有助于克服上述障碍(图1)。美国食品药品管理局(FDA)和医疗保险和补助服务中心(Centers for Medicare & Medicaid Services,CMS)目前在评估是否可针对分子诊断更为广泛地应用这些方案。本文评估了旨在促进分子诊断商业化进程的CED和平行审评的潜在适用性,并就如何加强这些创新方案提出建议。
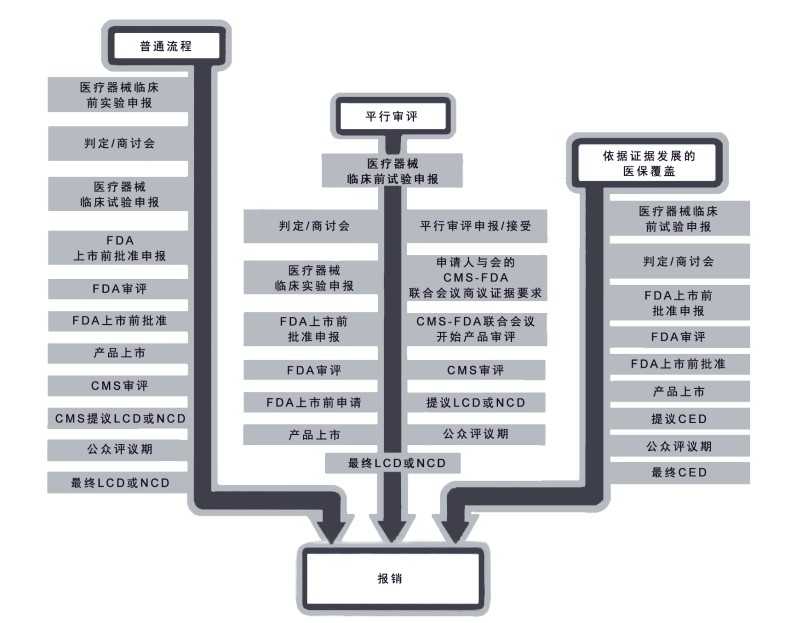
图为针对假设的诊断产品的FDA规定的批准流程和CMS流程。平行审评和CED能加快产品上市。
分子诊断:需要新对策
分子诊断包括能通过多种分子实体,如DNA、RNA和蛋白质,对疾病进行预测、诊断、预后和治疗的检测。FDA和CMS在这些检测的研发和商品化过程中都扮演了关键的角色。FDA规定作为医疗器械的诊断产品在上市前应提供“安全性和有效性的合理保证”,而在将产品纳入医保覆盖范围之前,CMS也承担有判定产品是否“合理和必要”的任务。这些法定授权看似有些交集,但在法律上是严格区分的,落实在实践中也非常不同。
FDA和CMS近来的政策变化为需要满足这两个机构要求的临床证据带来了不确定性。例如,FDA正在考虑针对实验室研发的检测产品(LDTs)(在单一实验室里研发和完成的诊断产品)的可能的新监管政策。同时,FDA也在修订其对于合作开发的医疗产品和伴随诊断产品的政策,正处于对广泛采用的510(k)审批程序(通过与现有设备的对比来审批医疗器械的上市)做出重大改变的阶段。同时,尽管CMS依旧凭借着相同的(但未明确定义的)“合理及必要”的准入门槛来界定医保范围,但是这些标准的执行似乎正在向比之前更有说服力和更具临床相关的有效性和患者结果数据的方向转变[1]。
这些转变中的依据证据的预期,营造出一种氛围,要求生成成本不断增长的临床依据来满足不同的监管标准,却没有为产品开发商带来可预测、透明和对满足新的证据标准所需的更大规模的研究进行投入的经济动因。此外,FDA的批准和CMS医保覆盖判定之间的延迟所招致的花费可成为这些医疗技术的临床应用和前景投资的重大阻碍。
分子诊断所面临的监管和医保政策困境的最终解决方案,也许需要法律或规章的彻底改变,而这一改变在当前的政治经济环境下不可能发生。与此同时,在当前的法律和规章框架下,渐进而具有价值的进展却是可能的。尤其是两个创新性的政策措施——CED和平行审评——为FDA和CMS减少创新医疗产品的评估过程中的关键瓶颈带来了希望。CED使CMS能够在产品申请人收集附加的临床依据知会CMS做出最终医保判定的前提下,将新的产品和服务临时性纳入医保报销覆盖范围。平行审评课使申请人在产品审评流程的早期即能与CMS和FDA举行会议,以明晰和同步协调这两个机构各自对证据的要求,并减少低效率和不一致的数据要求。
当前,扩大和加强CED和平行审评出现了契机,尤其是对于高价值的分子诊断产品。奥巴马政府制定的《国家生物经济蓝图》近期提议扩大这两项政策的应用,以促进生物医药创新[2]。美国国家医学研究院在近期举办的研讨会也强调了CED和平行审评及它们与分子诊断产品的关联[3]。
依据证据发展的医保覆盖
根据法律规定,美国医疗保险计划为医保受益人对用于诊断和治疗的“合理和必要的”的产品提供医保覆盖。CMS将能用于个体并反映疾病状况或症状的诊断服务和用于没有任何病征个体的预防或筛查服务进行了区分。在历史上,医疗保险已经为很多诊断服务提供覆盖,而限制对一些法律中有具体规定的筛查和预防服务提供覆盖。美国平价医疗法案修订了相关条例,允许CMS覆盖所有CMS认为“对疾病或伤残的预防或早期检测合理和必要”的和由美国预防医学工作组认可的预防服务都可以纳入医保,但尚不清楚这些条款将如何影响医疗保险覆盖的未来做法。
CMS和地方医疗保险管理订约机构共同承担判定特定产品是否符合“合理和必要”的准入门槛这一责任。CMS每年大约完成10–20项全国医保覆盖判定(NCDs),完成之前需经过法定的6个月到9个月的流程,包括系统性的证据评估、拟定决定公示、回应公众评论和发布最终决定。NCDs在全美范围内实施,并取代任何由订约机构作出的相矛盾的决定。区域订约机构完成每年绝大部分的医保覆盖决策,通过一个相似的但较宽松的流程。这些地方医保覆盖判定(LCDs)只适用于由订约机构监督的地理区域提供的产品和服务。然而,对于某些分子诊断产品,全国所有的检测都由一所实验室进行,因此被一个当地的订约机构评估和报销,这意味着这些用于这些检测的LCDs本质上就是NCDs。
CMS完成的NCDs可由多种方式来指导报销决定。偶尔,会为某种产品或服务的在所有情况下提供医保或在所有情况下拒绝医保覆盖。在一些案例中,推迟地方订约机构的决策。最为常见的是,NCD只在一些有可用证据的特定情形下提供医保。2006年,CMS正式提出了启用CED作为一种有条件医保覆盖的独立形式这一额外选项的纲要[4]。
CED使得CMS在产生追加的临床数据,更好地为机构的长期医保覆盖决策提供信息的情况下,为有前景的新技术提供临时报销。为接受对使用产品的医保报销,可能要求医疗服务提供方参与临床试验或登记以生成CMS要求的临床依据。在CED背景下开展的临床研究费用由CMS承担而不是由产品申报者承担。这样的机制能够帮助新产品申报者解决获取CMS要求的高质量临床证据所需的费用难题,同时也使CMS能够做出更加基于证据的长期医保覆盖判定,并使患者能够更早地使用到有前景的新技术。
自1995年起,CMS已经使用了18次CED模式的政策。第一次是评估治疗肺气肿的肺减容手术,并说明了该机制在改良CMS医保流程方面的潜力[5]。为应对不断增长的手术量和其有效性数据的匮乏,CMS通过一个多中心临床试验限制了这种手术的报销覆盖范围。这项研究显示了该手术只对一小部分患者有益,同时增大了其他患者的死亡率。通过使用这些数据,CMS将医保覆盖限制于对该手术确定临床效益的患者。这一方法,需要一次性花费3500万美元的研究费用,通过取消对无效干预的医保覆盖,每年估计为CMS节省1.5亿美元[6]。
CED在早期获得的成功,证明了这一方法在促进更多基于证据的CMS医保覆盖决策方面的潜力。然而,该政策仅有的两次应用,就导致了NCDs的修订,而其它的研究尚未开始或尚在进行中。认识到CED具有未能充分发挥的潜力,CMS在2011年宣布有意修订该政策,并于2012年1月征询公众意见[7]。2012年11月,CMS发布了CEDs新的指南草案,并于2013年初再次征询公众意见。根据《生物经济蓝图》,“CMS认为从CED初步实施中学到的经验教训,预示着其更为频繁的使用,并在更可靠地保证新技术能实现其最初宣称的优势的同时,为创新带来了可以预期的动因”[2]。
分子诊断为CED的应用提供了有前景的一类产品。CMS和其它支付方经常为新的分子诊断产品的报销要求临床数据,往往不同于FDA为这些产品的批准所要求的数据。CED提供了一个获取有关诊断检测价值的追加数据的有用路径,而不会阻碍有前景的创新产品的研发。例如,CMS近期针对基因型导向的华法林给药检测产品启用了CED[8]。
为扩大CED在分子诊断检测方面的应用,CMS应该解决下列瓶颈:
(1)明晰CED在补充FDA批准的上市后要求方面的功能。在判定一种分子诊断产品是安全和有效时,FDA有时会要求申请者收集批准后研究的追加数据。所要求的数据可能会与CMS为医保覆盖决策所要求的数据有所交集。针对批准后研究的基于CED的医保覆盖,将有助于确保确实开展了研究。继续实施这一选择之前,必须明晰将FDA批准后研究与CED研究相结合的机制和权威。
(2)明晰地方订约机构实行CED的规程和保障措施。地方订约机构完成大部分的医疗保险决策,而CED仅仅是针对NCDs设定的。地方订约机构是否具备完成CED判定的能力,引发了对花费更大、效率更低和一致性更差的医保模式的忧虑,尤其是当地方订约机构使用不同的方法和终点来处理同一种产品时。然而,某些分子诊断检测在全国只在一个独立的实验室内进行,并因此只由一家地方订约机构来评估和提供医保报销。CED对这些检测的应用,能够作为判定地方订约机构是否具备应用CED能力的试点案例。在广泛扩展至地方订约机构之前,必须设计出地方订约机构实施CED的适当保障措施。
(3)在简化NCD流程中应用CED。一项NCD的正常审评时间为9个月,这段延迟被产品申请者认为存在问题,他们期望CMS能紧随FDA的批准做出支付决定。在FDA批准3个月内公布CED决定,同时保留所需的30天征询公众意见期应该是可能的。这种简化的机制可能会使CED路线对新产品申请人更具吸引力,而不会影响申请人向CMS递交的数据质量。
平行审评
2011年10月,FDA和CMS发起了平行审评测试方案,目前正在按照这个方案分别为批准和医保评估医疗器械[9]。该测试方案预期计划运行两年,尽管这两个机构可以在方案实施测试阶段修改时间期限。该方案允许CMS在FDA审评新的器械同时开始其医保覆盖判定流程。这个自发的方案保证两机构各自的审评标准不变,同时在两个机构对数据的要求具有连续性而非一致时,寻求减少低效。正如生物经济蓝图所描绘的,“通过使CMS更早介入这一流程,预期平行审评能限制产品申报者和机构审评员的重复工作,减少新商品上市并从医保和其它提供方获得报销需要花费的时间”[2]。尽管一直以来公司在产品研发阶段都可以非正式地向CMS咨询,但平行审评试验计划扩展和规范FDA和CMS协调产品审评提供了机会[10]。
平行审评试验计划对医疗器械有严格限制,具体对符合以下标准之一的产品执行平行审评:(1)申请者有医疗器械临床前研究豁免(IDE)或已批准的IDE申请名称(寻求FDA开始临床试验批准的最初步骤);(2)需要为上市前批准(PMA)提交原始或补充申请或从头审评请愿书的新技术(FDA对于医疗器械对严格的流程);或(3)应列入A部分或B部分医疗保险福利类型范围内的或不属于NCD的新技术[10]。许多分子诊断产品都属于上述范围之一,并适合接受平行审评。与其它医疗仪器相比,FDA和CMS对分子诊断产品的数据要求通常更不确定,因此特别有利于使它们受益于合作审评和平行审评对数据的要求。
迄今为止,只有一种分子诊断产品(Cologuard,Exact Sciences)参与了平行审评路径(http://investor.exactsciences.com/2010AR/fda/index.html )。缺乏更广泛的参与可能源于平行审评的新奇性,业界可能认为这只不过是用一种不确定取代了另一种。此外,产品开发商同样在担忧从大量的私营支付方获取保险支付,可能不愿意将其数据收集狭隘地集中于CMS的要求上,尤其是在他们的产品设计是针对不恰好符合属于医疗保险范围内的类别的患者群体时。在这种情况下,产品研发人员可能会期望通过致力于与个别地方订约机构和私营支付方来协商以获得更有利的结果。最重要的是,一个早期的NCD否决判定,会摧毁一个产品商业上获得成功的所有前景。企业或许会不愿将他们的产品投注于这样一场“孤注一掷”的赌博。
平行审评试验计划的少数修订,可能会使该计划对分子诊断产品研发者更具吸引力,并为支付方、提供方和患者提供更快的使用机会和更为可靠的证据。这些变化包括:
(1) 重新考虑按照510(k)审批的分子诊断产品的资格。目前的试验计划不包括按照510(k)审批的器械,因为很多510(k)申请不包括实质性临床数据。此外,因为501(k)路径通常已经比PMA批准更快速,因此对于大多数医疗器械对平行审评的需求较少。然而,通过510(k)路径向FDA提交分子诊断产品,比其它产品更可能包括临床数据并花费更长的审批时间,针对这些产品的510(k)例外变得不太合理。风险投资家倾向于投资适合510(k)的诊断产品,而不是需要经过完整PMA的产品,显示出取消现行510(k)例外会增加风险资本支持的公司对该计划的兴趣。类似地,该计划应该足够灵活以适应未来对于诊断产品新的监管路径,比如正早讨论中的LDTs,以及针对未来的新技术,如全基因组测序的新技术路径。
(2)在平行审评的最初阶段取消NCD要求。试验计划有特定设计,能缩减FDA批准和CMS全国医保覆盖判定之间的时间。虽然对针对随后选择退出NCD的参与者的机制,但NCD是该计划设想的结果这一事实,可阻止担心遭到否决的开发商进入平行审评路径。如果CMS在自行发布NCD或允许地方订约机构发布他们自己的LCDs中保持中立,这一态度将减小开发商的风险,更多的诊断产品开发商将参与平行审评计划。即使产品申请人已经选择在产品研发阶段咨询CMS,许多新产品申请人目前花费大量时间和资源来寻求当地医保覆盖;因此,将平行审评与LCDs捆绑可减轻这一重负。
(3)为运用平行审评带来新动因。平行审评可能增大NCD被否决的风险这一观念可能阻碍产品开发商的参与。FDA和CMS可建立激励机制,以抵消这一忧虑,例如给予参与产品优先审评以加速其上市,或将审评与CED相联以支付收集数据的费用。由于两个机构在将何种产品纳入平行审评计划这一问题上有着慎重的考虑,他们可能会选择将上述激励对准显示出对于患者尤其有前景的产品。
即使平行审评试验计划每年仅限于三至五种器械,但可以为申请产品产生一个更高效的监管和医保报销流程,并为相关产品带来更加清晰的来自两大机构的证据需求。
对患者的承诺
分子诊断是体外诊断市场发展最快的部分。CED和平行审评提供了创新性的的监管和保险支付路径,能够加速、协调和促进这些产品的数据生成和临床应用。CMS和FDA正对这些计划重新评估,为其针对生物医药创新中这部分产品的应用开启了一扇机遇之窗。
参考文献和注释
[1] S. S. Dhruva, S. E. Phurrough, M. E. Salive, R. F. Redberg,CMS’s landmark decision on CT colonography—Examining the relevant data. N. Engl. J. Med. 360, 2699–2701(2009).
[2] U.S. White House, National Bioeconomy Blueprint (April 2012); www.whitehouse.gov/sites/default/files/microsites/ostp/national_bioeconomy_blueprint_april_2012.pdf.
[3] Genome-Based Diagnostics: Clarifying Pathways to Clinical Use—Workshop Summary (Institute of Medicine, Washington, DC., 2012); www.iom.edu/Reports/2012/Genome-Based-Diagnostics-Clarifying-Pathways-to-Clinical-Use.aspx.
[4] R. A. Lindor, Advancing evidence-based medicine by expanding coverage with evidence development. Jurimetrics 52, 209–237 (2012).
[5] M. R. Gillick, Medicare coverage for technological innovations—Time for new criteria? N. Engl. J. Med. 350, 2199–2203 (2004).
[6] S. R. Tunis, S. D. Pearson, Coverage options for promising technologies: Medicare's 'coverage with evidence development'. Health Af . 25, 1218–1230 (2006).
[7] CMS, CED Public Solicitation (7 November 2011): www.cms.gov/medicare-coverage-database/details/medicare-coverage-document-details.aspx?MCDId=8&McdName=CED+Public+Solicitation&mcdtypename=Guidance+Documents&MCDIndexType=1&bc=AgAEAAAAAAAA&.
[8] CMS, National Coverage Determination (NCD) for pharmacogenomic testing for warfarin response (90.1)(5 April 2010); www.cms.gov/Medicare/Medicare-Contracting/ContractorLearningResources/downloads/JA6715.pdf.
[9] CMS and FDA, Pilot program for parallel review of medical products, 76 Fed. Reg. 62808-62810 (11 October 2011).
[10] D. A. Messner, S. R. Tunis, Current and future state of FDA-CMS parallel reviews. Clin. Pharm. Therapeut. 91, 383–385 (2012).