|
首页
>
资讯
>
国内药政每周导读:2021年度审评报告发布,CDE连发细胞与基因治疗CMC技术指南,药包材迎来GMP草案
出自识林
国内药政每周导读:2021年度审评报告发布,CDE连发细胞与基因治疗CMC技术指南,药包材迎来GMP草案
2022-06-06
【创新研发】
5.30,CDE发布《局部给药局部起效药物临床试验技术指导原则》
由“版本历史”可知,本文曾于2021年11月征求意见。
局部给药局部起效药物(locally applied, locally acting products,LALAP),是指应用于局部并在应用部位发挥作用的药物。此类药物如出现全身作用,则被认为是非预期的药物作用。
与系统给药药物相比,局部给药局部起效药物在处方组成、剂型特点、给药途径等方面具有特殊性,因此,应针对性进行临床试验设计和评价,包括局部给药后局部和全身的耐受性、安全性、局部和系统药代动力学、局部药效动力学、剂量探索等。
对于这类正式文件,识林建议企业用户登录后,结合“页面比对”工具,制作花脸稿,理解CDE在修订时体现出的科学考量和监管导向。
此外,CDE连续发布了下列几份正式指南。
5.31,CDE发布《体外基因修饰系统药学研究与评价技术指导原则(试行)》
由“版本历史”可知,本文曾于2020年9月征求意见,当时的名称还是《基因转导与修饰系统药学研究与评价技术指导原则(征求意见稿)》。
体外基因修饰系统,本身并非药品,而是在人体外,采用基因工程技术构建的修饰系统,可有效地将遗传物质等转入特定目的细胞,用于修饰目的细胞的遗传物质、改变基因表达方式或调节细胞生物特性等。
目前,慢病毒载体、γ-逆转录病毒载体等常见用于将嵌合抗原受体(Chimeric Antigen Receptor, CAR)基因导入T细胞,以实现CAR-T细胞对肿瘤的靶向杀伤;游离型载体(Episomal Vector)、仙台病毒载体等可用于将转录因子导入细胞,通过重编程获得诱导多能干细胞,为其衍生细胞产品的生产提供起始原材料。将来,预计会有更多样的载体设计适用于不同类型的产品。
基因修饰系统种类多样,载体设计、制备过程以及质量控制等方面的差异直接影响到最终产品的安全性和有效性,且其来源可能不同,质量管理体系存在差异。为保证基因修饰系统质量符合临床应用的要求,需对其进行充分的质量研究。因此,有必要细化不同类型基因修饰系统药学研究的技术要求。
5.31,CDE发布《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》
由“版本历史”可知,本文曾于2020年9月征求意见。
本指导原则中免疫细胞治疗产品是指源自人体(自体/异体)细胞或人源细胞系的细胞,经过体外操作,包括但不限于分离、纯化、培养、扩增、诱导分化、活化、遗传修饰、细胞库(系)的建立、冻存复苏等,再输入或植入到患者体内,通过诱导、增强或抑制机体的免疫功能而治疗疾病的免疫细胞治疗产品,例如嵌合抗原受体T细胞(Chimeric Antigen Receptor T-Cell,CAR-T)、树突状细胞(Dendritic Cell,DC)等。
胰岛细胞、软骨细胞等体细胞,以及细胞与非细胞成分的组合产品的细胞部分也可以参考本指导原则。细胞衍生产品,如细胞外泌体、细胞裂解物、灭活细胞等产品,其细胞部分的药学研究也可能适用。
对于经基因修饰的免疫细胞治疗产品(如CAR-T等),其细胞部分可以参考本指导原则,基因修饰部分可以参考其他相关技术指南(如同期发布的《体外基因修饰系统药学研究与评价技术指导原则(试行)》)。
本指导原则不适用于干细胞、输血或移植用的造血干细胞、生殖细胞,以及由细胞组成的类组织、类器官产品等。本指导原则适用于按照药品管理相关法规进行研发和注册申报的免疫细胞治疗产品,主要适用于上市申请阶段的药学研究。
5.31,CDE发布《体内基因治疗产品药学研究与评价技术指导原则(试行)》
由“版本历史”可知,本文曾于2020年9月征求意见。当时的名称还是《基因治疗产品药学研究与评价技术指导原则(征求意见稿)》
之所以更名的原因,在于定义的进一步明确。
体内基因治疗产品将外源基因(或基因编辑工具)通过适当的载体直接导入人体发挥治疗作用,而体外基因治疗产品一般在体外将外源基因(或基因编辑工具)导入细胞,制备成为经基因修饰的细胞或细胞衍生产品,最终经回输以发挥治疗作用。
由于体内和体外基因治疗产品在产品类型、基因载体类型与设计、载体的靶向性需求、起始原材料的管理、产品的纯度、杂质水平的控制、生产模式和质量风险等方面存在一定差异,因此,两类产品在研发和技术要求方面存在一定的差异,有必要进行分类规范。
不过,在《基因治疗产品非临床研究与评价技术指导原则(试行)》(CDE,20211203)和《基因修饰细胞治疗产品非临床研究技术指导原则(试行)》(CDE,20211203)这类已经发布的指南中,所谓“基因治疗产品”和“基因修饰细胞治疗产品”,与本文中“体内”和“体外”,以及同期发布的指南中“体外基因修饰系统”,这些概念之间的异同,还需进一步厘清。
企业用户可登录识林阅读主题词【基因治疗】和【细胞治疗】,全面了解这两类产品的基础知识和监管体系。
【CMC与仿制药】
5.31,药典委征集《红外光谱集》(第一卷至第五卷)执行意见
药典委拟编制出版《红外光谱集》(2022年版),内容主要包括1995年至2005年已出版《红外光谱集》(第一卷至第五卷)的品种及《中国药典》2020年版二部新增原料药品种的红外光谱。为确保新版《红外光谱集》的质量,现对《红外光谱集》第一卷至第五卷执行过程中可能存在的问题公开征求社会各界意见。
6.1,药典委发布新的辅料标准草案公示
包括:
识林用户可登录APP和PC查阅中国药典以及标准公示数据库。
【注册,审评,审批】
6.1,NMPA发布《2021年度药品审评报告》
NMPA与CDE在2021年成果颇丰。
报告中有大量数据,读者可自行查阅。比数据更为实用的,是提炼的信息与结论,以及其中体现出的导向与趋势。
——CDE的工作效率越来越高,但工作量也越来越大。47个创新药批准,再创新高,按时限完成率取得历史性突破。当然,受理数量也大幅度增长。
——要事优先,审评资源聚焦临床急需,成效显著。四条快速通道(突破性治疗,附条件批准,优先审评审批,以及特别审批程序)运行良好。临床急需境外新药,81个品种中,已有54个提出申请,51个获批上市,且100%在规定时限内完成NDA审评。新冠病毒疫苗和药物均被纳入专用通道。
——更高的监管透明度,更低的企业风险与成本,企业喜闻乐见。沟通会议425次,大幅增长58.58%,其中II类会议(即为药物在研发关键阶段而召开的会议)占比70.35%。
——中药注册审评路径逐渐走通,曙光初现。批准(NDA14件,IND34件)和申请(NDA14件,IND34件)均显著增长。
——指南越多越细,审评标准越明确,但也越高。ICH实施53个,比例达到84.13%。NMPA总计发布技术指导原则87个,总数达到361个。
【生产质量管理】
6.2,NMPA征求《药包材生产质量管理规范》意见
“原辅包”三者一向并称。
其中,原料药作为药品管理,原料药GMP是中国GMP官方附录之一,也在中国GMP指南占据一席之地。
辅料GMP也已发布,不过还是早在2006年3月发布的《药用辅料生产质量管理规范》。
本文发布,意味着药包材终于迎来自己专属的GMP。
阅读本文,可见其整体架构与药品类GMP颇为类似,质量体系、机构人员、偏差变更等等各种要素齐备,当然,许多要求有一定简化。
读者可关注下列要点:
——适用范围。本规范中的药包材,主要是指与药品直接接触的包装材料和容器,也包括功能性次级包装材料、表面印刷材料、组件和给药装置等。
——强调内部审核与管理评审。企业应当制定内部审核与管理评审管理规程,明确内部审核和管理评审的方式和标准。这一内容放在第二节,共计四条,颇为突出。
——第十三章专门讲“用户管理服务”。毕竟药包材厂家是药企的供应商,而药品才是监管的最重点,因此药包材GMP的要求需要符合其在产业链的定位。
征求意见截至7月2日。
【新药批准和报产】
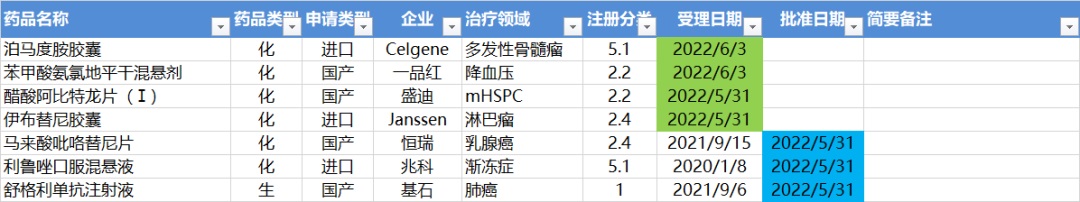
5.30-6.5,NMPA发布3个新药批准,CDE受理4个新药上市
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
作者:识林-实木
识林®版权所有,未经许可不得转载。
|