|
首页
>
资讯
>
国内药政每周导读:Q9(R1)中文版,eCTD实施指南,Q3D(R2)与M10适用,参比目录第64批,江苏GVP实操指南
出自识林
国内药政每周导读:Q9(R1)中文版,eCTD实施指南,Q3D(R2)与M10适用,参比目录第64批,江苏GVP实操指南
2023-02-06
【CMC与仿制药】
1.29,【NMPA】关于适用《Q3D(R2):元素杂质》《M10:生物分析方法验证及样品分析》国际人用药品注册技术协调会指导原则的公告(2023年第16号)
申请人需在现行药学研究技术要求基础上,按照Q3D(R2)指导原则的要求开展研究;自2023年7月29日起开始的相关研究(以试验记录时间点为准),均适用Q3D(R2)指导原则,Q3D(R1)指导原则同时停止实施。
申请人需按照M10指导原则的要求开展研究;自2023年7月29日起开始的相关研究(以生物样品分析原始记录时间点为准),均适用M10指导原则。
识林曾对比Q3D(R2)与(R1)的变化,见“ICH Q3D(R2)正式版发布:部分 PDE,各论,皮肤与透皮给药元素限度”。
1.31,【NMPA】关于发布仿制药参比制剂目录(第六十四批)的通告(2023年第6号)
目前,国内参比制剂目录已发布六十四批,公示六十六批。
见识林“参比制剂数据库”或专题页面“中国化学仿制药参比制剂目录公示批列表”。
2.01,【药典会】新增6篇通则、3篇中药、6篇辅料标准草案的公示,转发48个中药配方颗粒国家药品标准
新增的通则包括:
其他内容可查阅识林专题页面“国家药典委员会”,或“中国药典数据库”。
【注册、审评、审批】
1.30,【CDE】关于公开征求《eCTD实施指南V1.1(征求意见稿)》及《eCTD验证标准V1.1(征求意见稿)》意见的通知
为配合2023年1月1日实施的注册申请电子申报,全面进入电子申报时代,CDE及时修订了eCTD的主要文件。
(一)《eCTD 实施指南 V1.0》 修订内容
删除 2.8 章节关于纸质资料的递交要求,删除 4.1.2 章节和附件说明函中关于纸质资料的相关内容,包括“关于纸质资料与 eCTD 申报资料内容一致的承诺”和“关于按规定时限一次性提交全部纸质申报资料的承诺”,修改 7.6 章节关于文件大小的要求,将单个 PDF 文件应控制在 500MB 以内修改为 200MB。
(二)《eCTD 验证标准 V1.0》 修订内容
修改验证标准第 2.2 条,将验证说明由“超出允许大小的文件会提示警告信息。普通单个文件最大允许 500MB,单个 SAS XPT 文件可支持 4GB。”调整为“超出允许大小的文件会提示警告信息。普通单个文件最大允许 200MB,单个SAS XPT 文件可支持 4GB。”
【GMP与检查】
1.10,【上海医药商业行业协会】临床试验用药品供应链管理规范
临床试验用药品供应链突破了传统对药物注册、生产、流通、使用的明确界限,各相关方过程管理尚处于标准空白区,没有明确管理要求。
国家局于2022年发布了《临床试验用药品GMP附录》,但重点关注在对临床试验用药的生产、检验等环节。本文件来自行业协会,对法规未明确的供应链环节管理,尤其是采取第三方服务方式的供应链管理环节,明确了各相关方的职责划分、软硬件条件、操作规范等管理要求。
起草单位包括上海局药品审评核查中心,以及上药、国药、辉瑞等,还包括复旦大学附属中山医院等用药单位。
行业协会的指南,可以作为法规指南的补充,具有相当参考价值。见识林专题页面“行业协会” 。
1.30,【CFDI】关于公开征求ICH指导原则《Q9(R1):质量风险管理》中文翻译稿意见的通知
距离ICH于1月20日发布Q9(R1)定稿指南仅过去10天,CDE就发布中文翻译稿。
定稿指南更新了已有 18 年历史的原指南,涵盖了 QRM 的原则、一般 QRM 流程、风险管理方法、QRM 与行业和监管运营的整合。定稿指南还有两个附录,包括 QRM 方法和工具以及 QRM 的潜在应用。
事实上,定稿指南与 2021 年 12 月发布的第 2 阶段草案相比几乎没有变化。识林企业用户可登录查阅识林的对比解读文章“ICH Q9 step 2 比对总结” 。
尽管如此,定稿就意味着开始正式实施,如何在药品生命周期体现质量风险管理,不仅质量部,其他各个部门都需要学习并思考。
更多信息,可阅读资讯“ICH Q9(R1) 质量风险管理修订指南正式定稿”。
【政策法规综合】
1.28,【江苏省】关于发布《江苏省药品上市许可持有人药物警戒质量管理规范实操指南(试行)》的公告
本指南的主要依据不仅仅是国内的GVP《药物警戒质量管理规范》,此外还参考了欧盟药物警戒质量管理规范和国内的相关技术性文件(指南/指导原则),如《欧盟药物警戒质量管理规范》(国家药品监督管理局药品评价中心,天津科技翻译出版有限公司)、《药物警戒检查指导原则》、《药物警戒体系主文件撰写指南》、《个例药品不良反应收集和报告指导原则》等。
本文称为“实操指南”,特点是采用表格的形式,将“实操指南”和GVP原文,以及GVP检查指导原则的原文一一对应起来。文末还有3个流程图附件,包括药品不良报告,信号检测,以及药物聚集性事件处理。
这种形式颇为少见,更便于企业理解和运用。江苏省外企业亦可参考。
【新药批准和报产】
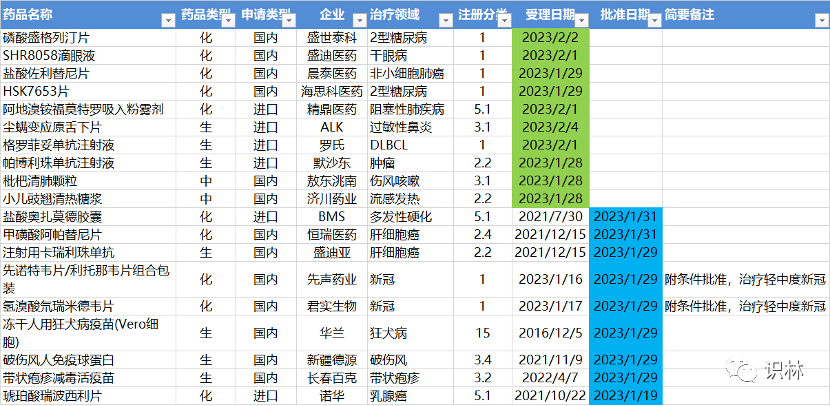
1.28-2.05,NMPA发布9个新药批准,CDE受理10个NDA
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林-实木
识林®版权所有,未经许可不得转载
|