|
首页
>
资讯
>
国际药政每周概要:Q3D修订定稿,FDA 药学审评新指南,EP制剂各论和肝素标准品,EMA植物药质量,国会调查新冠疫苗GMP
出自识林
国际药政每周概要:Q3D修订定稿,FDA 药学审评新指南,EP制剂各论和肝素标准品,EMA植物药质量,国会调查新冠疫苗GMP
2022-05-17
【创新药物研发】
5.09,【FDA】指南议程:2022日历年 CBER 计划发布的指南(2022年5月更新)
CBER 公布2022日历年指南发布计划,完整翻译如下:
- 类别--血液和血液成分:
-
-
-
-
- 关于血液和血液成分捐赠适用性、捐赠者资格和源血浆隔离保留要求的合规政策;行业指南草案
-
- 降低通过血液和血液成分传播克雅氏病和变异克雅氏病的可能风险的建议;行业指南
-
- 类别--组织和先进疗法:
-
-
-
- 人体细胞、组织以及基于细胞和组织的产品 (HCT/Ps) 的监管 - 对小型组织的合规指南;行业指南
- 再生医学疗法自愿共识标准认可计划;行业和内部员工指南草案
- 关于确定人类细胞、组织以及细胞和组织产品 (HCT/Ps) 供体资格的建议;行业指南草案
- 人类细胞和基因治疗产品的生产变更和可比性;行业指南草案
- 类别 – 疫苗:
-
- 关于使用粪便微生物群移植治疗对标准疗法无反应的艰难梭菌感染的研究性新药要求的执行政策;行业指南
5.10,【FDA】CDER 的加速罕见病治疗(ARC)计划
CDER 发起加快罕见病治疗(Accelerating Rare disease Cures,ARC)计划。
该计划将充分调动 CDER 的专家资源,采取各种行动,从战略高度对 CDER 的罕见病工作进行管理和协调。
ARC 的愿景(Vision):
- 加快并增加有效且安全的治疗选择的开发,应对未满足的罕见病患者的需求。
ARC 的使命(Mission):
- 通过科学和监管的创新与保证,加速罕见病患者的治疗可及性。
文中还罗列了接下来的重要活动,近期批准的药物等等。
5.12,【WHO】WHO 和 MPP 宣布与 NIH 就 COVID-19 卫生技术达成协议
世卫组织 COVID-19 技术访问池 (C-TAP) 和药品专利池 (MPP) 今天与美国国立卫生研究院 (NIH) 敲定了一项许可协议,用于开发 COVID-19 相关的几种创新疗法、早期疫苗和诊断工具。
这些透明、全球性和非排他性的许可证将允许来自世界各地的制造商与 MPP 和 C-TAP 合作,使生活在低收入和中等收入国家的人们能够使用这些技术,终结新冠疫情。
在两个许可证下提供了11种 COVID-19 技术,包括目前可用的 COVID-19 疫苗中使用的稳定刺突蛋白、疫苗研究工具、治疗和诊断开发以及早期候选疫苗和诊断。
【注册、审评、审批】
5.09,【FDA】指南草案 产品质量评估的风险及获益的考虑
本指南草案是描述 FDA 在审评 NDA、BLA 等申报资料中与 CMC(药学)部分内容时采取的获益-风险原则。
从内容上看,是 FDA 对创新药 CMC 审评尺度的一次阐述,相当重要。
FDA 探讨了如何评估产品质量相关问题的风险、不确定性来源和可能的缓解策略,以及 FDA 如何理解这些问题对产品的潜在影响。
产品质量评估确定申请人的产品研发、生产工艺和控制策略在申报设施中开展生产时,将始终如一地产出质量合格的产品。
当做出有关批准 NDA 或 BLA 的监管决定时,FDA 会考虑产品相关的总体利益和风险,包括与未解决的产品质量问题相关的任何残余风险。本指南还讨论了如何在监管决策的背景下如何处理未解决的产品质量问题。
5.12,【FDA】指南更新SOPP 8417:实施和管理风险评估与减轻策略
本指南上一版本还是2020年12月,用于 CBER 内部员工。
与 CBER 打交道的国内药企应关注本文,预判提交风险评估与减轻策略(REMS)的必要性。
回顾一下风险评估与减轻策略(REMS)的背景:
- 由 FDAAA(《食品药品监督管理修正案》)增补并随后由 FDASIA (《食品药品监督管理创新法案》)修订的《联邦食品、药品和化妆品 (FD&C) 法案》第 505-1 节授权 FDA 向企业索取 REMS,前提是 FDA 认为有必要提交 REMS 以确保对应药物(药品/生物制品)的收益大于风险。
FDA 可以要求申请人提交 REMS,负责决策的部门是负责审评药物的办公室(即 CBER 的药品办公室)和负责药物批准后安全的办公室(即 CBER 的生物统计和药物警戒办公室)。
5.12,【FDA】SOPP 8301:主文件的接收和处理
国内企业非常熟悉 CDER 的 MF 类文件,尤其是 DMF,而这份指南则专门用于 CBER 的 MF 类文件。
CBER 将 MF 分为4种类型:
- 1,Type II:原料药、原料药中间体、制剂及其制备中使用的物料或制剂的信息;
- 2,Type III:包装材料;
- 3,Type IV:关于辅料、着色剂、香精、香精或制备所用物料的信息;和
- 4,Type V:FDA 接受的参考信息,不属于 II 类到 IV 类。例如:
-
-
- 合同包装、制造、测试、灭菌等(包括有关在设施中制造/加工的其他医疗产品的信息)
- 所有器械主文件都归类为 V 型 MF。器械主文件可能包含有关医疗器械制造、加工或包装中使用的特定制造设施、流程、方法或组件的详细信息。他们还可以提供有关医疗器械成品的信息。
其他动态
5.09,【PMDA】再生医学产品 审评报告 新增 Ocural
5.09,【PMDA】药品 审评报告 新增 Paxlovid
5.11,【FDA】COVID-19 药品和非疫苗生物制品紧急使用授权 页面更新
5.12,【PMDA】药品 审评报告 新增 Actemra
5.12,【FDA】批准依达拉奉口服制剂用于治疗成人肌萎缩侧索硬化症 (ALS)
【cGMP与全球检查】
5.10,【美国国会】Emergent BioSolutions 的冠状病毒疫苗生产失败调查报告
这周二,美国国会公布了对位于美国马里兰州巴尔的摩市的新冠疫苗委托生产商 Emergent BioSolutions GMP 违规的《调查报告》【可登录识林阅读】。虽然报告本身不乏美国国内党派争斗的色彩,但有一些值得我国企业注意的地方。尤其是,企业内部、及与合作方和咨询师的往来邮件和文件,都可被传证并公开。
请见专题文章:【周末杂谈】国会对疫苗合同生产商 GMP 违规的调查报告
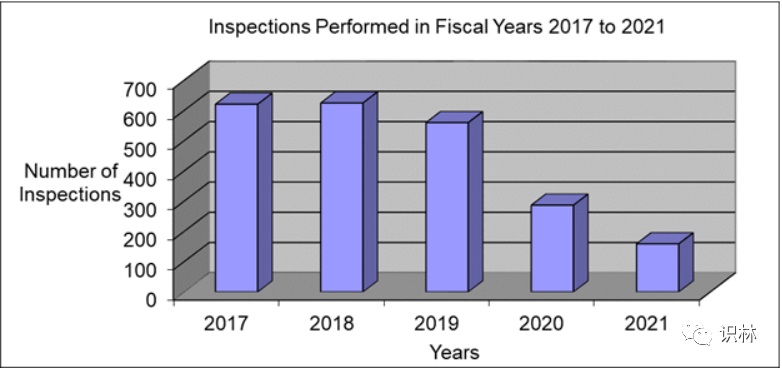
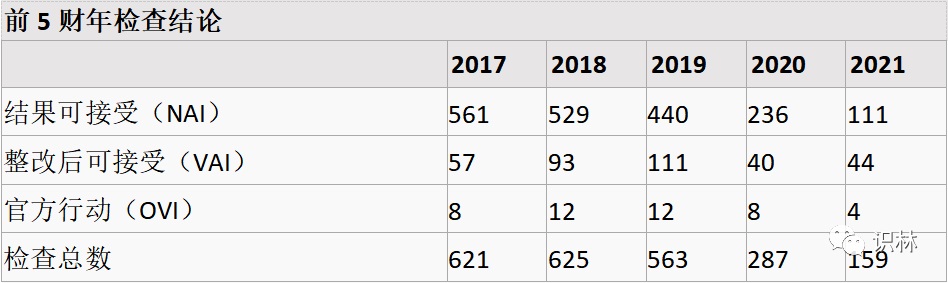
5.13,【FDA】HCT/P(人体细胞和组织相关产品) 检查信息(2021财年)
上图可见,检查数量持续下降。
上表可见,检查虽然下降,但 VAI 和 OVI 的比例在上升。
5.13,【EMA】关于质量受权人本人在场和个人住处征求意见
5月13日,EMA 发布题为“关于质量受权人本人在场和个人住处征求意见”的文件,征求意见的时间为2022年5月13日-6月13日,该文件提供了关于质量受权人(Qualified Person,QP)远程认证或确认的问答,内容包括:
其他动态
5.04,【EDQM】关于灭活 COVID-19 疫苗的新 OCABR(官方批签发) 指南
5.09,【加拿大】检查追踪 新增 印度 Bharat Biotech International Limited
5.10,【FDA】警告信 韩国 David Cosmetic Co., Ltd.
5.10,【FDA】警告信 美国 Brigham and Women's Hospital Inc.
5.13,【FDA】483 印度 Sun Pharmaceutical Industries Ltd.
5.13,【FDA】483 美国 Ark Pharmacy, PC (dba Regency Medical Pharmacy )
【CMC与仿制药】
5.05,【EDQM】开发含有化学定义的活性物质水合物或溶剂合物的药品各论的新政策
在 2022 年 3 月的第 172 届会议上,欧洲药典 (Ph. Eur.) 委员会批准了一项新政策,用于制定含有化学定义活性物质的水合物或溶剂化物形式的药品专论。
该政策区分了以水为溶剂分子的水合物和其他涉及有机溶剂的溶剂化物(以下称为“溶剂化物”)。
这项新政策有几个优点:
- 提升明确性,因为各论的范围将在标题中指示(无需阅读定义即可了解范围);
- 在活性物质和药品各论的开发和修订方面具有更大的灵活性:当含有新活性物质溶剂化物的药品获得批准时,将开发新专论,而不是修订现有各论。
5.05,【EDQM】细化包含化学原料药药品各论的技术指南
这是一篇 EDQM 对于化药制剂各论结构和内容的导读式指南。
药品各论的整体结构与已确证化学结构的物质各论的整体结构相同。
本指南阐述了与含有已确证化学结构的活性物质的药品相关的具体要点,这些要点未一般性指南中明确。
关于放射性药物制剂的专着不在本指南的范围内。
有关药品的各论应与 Ph. Eur 的通则一起阅读。除非特别豁免,相关总论的要求,例如剂型或药物制剂总论 (2619),适用于各论。
5.09,【ICH】Q3D(R2) 元素杂质指南正式发布
国际人用药品注册技术协调会(ICH)颁布了元素杂质指导原则修订版 Q3D(R2),是之前于2020年9月25日发布的Q3D(R2)草案(Draft)的最终版本(Final)。
此次修订版修正了附录2中金(Au)、银(Ag)和镍(Ni)的每日允许暴露量(Permitted/Permissible Daily Exposure,PDE),以及附录3中金(Au)和银(Ag)的各论,并增加了附录5:皮肤和透皮给药途径的元素杂质限度【相关资讯:ICH Q3D(R2)正式版发布:部分 PDE,各论,皮肤与透皮给药元素限度】。
识林将会专门翻译修订的内容,供用户参考。
5.10,【USP】美国药典-国家处方集 (USP–NF)大量更新,最新 PF 双月刊发布
更新内容主要有:
- 药典论坛(Pharmacopeial Forum,PF)最新双月刊发布
- 包括
- 22个 USP 药品各论修订,征求意见至5月31日;
-
-
- 修订公告(Revision Bulletins,RBs)
-
-
- 勘误(Errata,ERR)
-
-
-
-
-
企业用户可至识林“美国药典-国家处方集 (USP–NF)”页面检索,并跳转至 USP 官方页面。
5.10,【EDQM】生物标准化计划研究新出版物:低分子量肝素分析生物标准品
一篇关于建立第 11 批欧洲药典 (Ph. Eur.) 肝素低分子量测定生物标准品配制的生物标准化计划 (BSP) 研究结果的文章现已可在线阅读。
该文章已发表在 Pharmeuropa Bio & Scientific Notes 并被 Medline/PubMed® 引用。它提供了有关标准品配制、其建立方法以及统计分析的细节和结果的有用信息。
Pharmeur Bio Sci Notes 是 Medline/PubMed® 上引用的免费在线版本,可从欧洲药品和医疗保健质量局 (EDQM) 的网站公开访问。
5.12,【EMA】植物药/传统植物药质量指南
5.12,【EMA】质量标准:植物原料、植物制品和植物药/传统植物药的检验规程和可接受标准指南
EMA 连发两份植物药领域的重要指南,距离上一版本已过去近4年。
从标题即可看出,这两篇指南的主题都是产品质量,其中前者是概述,后者则是具体阐述一份植物药质量标准里的检测项目和制定依据。
目前,欧洲植物药市场广阔,获得广泛认可,美国也已开始批准植物药(相关资讯【欧盟植物药2022监管规划:草药专论,法规指南,审评程序】和【大疱性表皮松解症治疗药桦树皮提取物有望成为 FDA 批准的第三款植物药】等)。
尽管植物药并非中医理论指导的药品,但从其工艺和质量来看,国内中药企业具有相当的技术积累和供应链资源,也许能在植物药领域有所建树。
其他动态
5.12,【WHO】修订 多替拉韦 BE 指南
5.12,【FDA】仿制药计划月度绩效活动报告(2022财年)更新
5.13,【FDA】ANDA 类的 RS 对应的 RLD 列表更新
【监管综合】
5.06,【WHO】发布首份全球感染预防和控制报告
该报告表明,良好的 IPC 计划可以将医疗保健感染减少 70%。见 相关资讯【WHO 发布首份全球感染预防和控制(IPC)报告】
COVID-19 大流行和最近的其他大型疾病暴发凸显了卫生保健机构的管理方式(尤其是对感染预防和控制 (IPC) 不够重视)在多大程度上助长了感染的传播,从而对患者、卫生工作者和访客造成伤害。而世界卫生组织 (WHO) 的一份新报告显示,如果遵循良好的手部卫生和其他具有成本效益的做法,则可以预防 70% 的感染。
其他动态
5.10,【MHRA】合规监测流程(第2部分)——合规监测任务和申请流程
5.11,【EMA】危机准备时建立“主要治疗分组”列表和重大事件和/或公共安全紧急情况中建立“关键药物”列表的方法
5.13,【FDA】SOPP 8101.2:行业贸易组织联络会议的安排和记录
作者:识林-实木
识林®版权所有,未经许可不得转载。
|