|
首页
>
资讯
>
国内药政每周导读:CGT临床沟通交流,血液制品GMP附录拟修订,Q7A问答中文,PV委托管理团体标准,第三批鼓励仿制目录
出自识林
国内药政每周导读:CGT临床沟通交流,血液制品GMP附录拟修订,Q7A问答中文,PV委托管理团体标准,第三批鼓励仿制目录
2024-01-01
本周生效的重点法规指南
上周国内药政导读
【CMC与仿制药】
12.25,【CFDI】关于公开征求《ICH Q7 原料药的药品生产质量管理规范指南问答》中文翻译稿意见的通知
征求意见截至2024年1月6日。
Q7A是国内原料药厂耳熟能详的ICH指南文件了,此次征求意见的是Q7A问答的中文翻译稿。
查阅“版本历史”,Q7A正文中文翻译稿于2020年12月征求意见,但迄今为止还未发文正式“适用”。
ICH页面可知,Q7A与问答分别于2000年11月和2015年6月发布,此后均无修订。
12.25,【卫健委】关于印发第三批鼓励仿制药品目录的通知
继10月份公示后,卫健委拟定《第三批鼓励仿制药品目录》(以下简称第三批目录),收录39个品种,涉及75个品规、13种剂型,覆盖抗肿瘤药及免疫调节剂、抗感染用药、神经系统用药、放射性诊断剂、心血管系统用药等12个方面治疗用药。
卫健委的解读中,指出前两批鼓励仿制药品(共计50个品种)颇有成效:
- 33个品种已获批上市,覆盖抗感染用药、抗肿瘤药及免疫调节剂、神经系统用药等15个方面治疗用药,其中包含7个罕见病用药。
- 形成充分市场竞争,保障药品供应。前2批目录已上市的33个品种中,3家及以上企业生产的品种15个,2家企业生产的品种6个,1家企业生产的品种12个;目录发布前已有上市药品,发布后有新增生产企业的品种9个。
- 药企得到实惠。前2批目录中,已有14个药品通过纳入优先审评审批程序予以上市许可,已有12个品种通过谈判进入国家医保目录。
12.26,【NMPA】关于发布仿制药参比制剂目录(第七十四批)的通告(2023年第67号)
截至目前,NMPA已发布参比制剂目录74批,CDE已公示77批。
识林用户可登录“中国参比制剂库”查阅。
12.28,【药典会】新增多篇化药、生物制品、中药标准草案的公示
具体请查阅识林“药典会标准公示”页面。
其中适用面较广的有:
其中,ADC总论重点阐述抗体偶联药物和其组成部分 ( 抗体、有效载荷、连接子或有效载荷-连接子)的相关要求,涉及抗体的生产和质量控制。对目前非常热门的ADC的CMC药学研究和生产质量管理都有重要指导意义。
中国药典针对化学药提供质量标准限度和分析方法,但对于生物制品,会通过总论和各论阐述具体产品的生产过程,这应算是生物制品一大特色。为何有此区别?建议感兴趣的识林用户阅读《为生物制品注册人辨析“制造检定规程”的来龙去脉》。
【注册审评】
12.29,【CDE】关于发布《细胞和基因治疗产品临床相关沟通交流技术指导原则》的通告(2023年第60号)
本文曾于2023年7月征求意见。正式文即日起实施,虽名为沟通交流,但有许多技术内容指导研发和临床。
通过“页面比对”制作花脸稿,正式文包含下列修订内容:
- 增加“拟申请附条件批准上市的,药品上市许可申请递交前,申请人应当讨论附条件批准上市的条件和上市后继续完成的研究工作等。”
- 增加“一般在早期探索性临床试验中的主要研究目的是探索产品安全性,则主要终点指标设置为安全性观察指标等。”
- 增加“参考同类产品...正在进行的已有初步数据的临床研究...”,而不只是“已完成”的临床研究数据。
以上仅为部分,建议企业用户利用“页面比对”制作花脸稿仔细阅读修订内容,进一步理解监管导向。
【GMP与检查】
12.26,【NMPA】公开征求《药品生产质量管理规范》血液制品附录(修订稿征求意见稿)意见
国家药监局组织对《药品生产质量管理规范》血液制品附录进行了修订,具体情况如下:
一、本次修订内容主要是增加对血液制品生产、检验环节信息化和可视化的相关要求。
二、修订内容包括:
(一)第二十五条增加了要求血液制品生产企业督促浆站采用信息化手段如实记录原料血浆采集、贮存、运输及检验数据的规定。
(二)参照生物制品附录中疫苗生产及检验信息化记录的要求,新增第三十五条。同时还新增了基于风险对关键生产、检验环节采取必要的可视化监控措施的要求,原第三十五条改为第三十六条。
三、本次修订工作,组建了由国家药品检查尖兵“百人计划”的专家组成的血液制品附录修订小组,经认真讨论研究并汇总专家组意见建议,形成了修订稿。
【政策法规综合】
12.26,【上海医药行业协会】药物警戒活动委托质量管理规范
又一份大家喜闻乐见的团标发布。
本文件规定了药物警戒委托的程序和要求,界定了委托内容、委托质量目标等。本文件适用于临床试验申办者、药品上市许可持有人持有人委托药物警戒产品和服务供应商开展药物警戒活动,同时适用于集团公司内部(含集团内商业公司)药物警戒活动委托。
回想2022年12月发布,2023年3月1日生效的《药品上市许可持有人落实药品质量安全主体责任监督管理规定》,从征求意见稿中删去了“药物警戒负责人等关键岗位人员...应为企业全职人员”的要求,改为“企业负责人...负责配备或指定药物警戒负责人”。但第二十条仍然规定,“持有人应当建立药物警戒体系,设立专门的药物警戒部门,按照药物警戒质量管理规范等要求开展药物警戒工作,进行药品不良反应及其他与用药有关的有害反应监测、识别、评估和控制等活动,最大限度地降低药品安全风险。”
至于PV可以外包的依据,则见于GVP《药物警戒质量管理规范》(2021.5)第三节,委托管理。
因此,MAH仍需建立PV体系,但PV工作可以外包。
相信B证企业应该会充分利用GVP外包,并从这份团标中获益。
团标尽管非强制,但也是标准体系的组成部分(国标、地标、行标、企标),并且这份团标由由上海市药品和医疗器械不良反应监测中心提出并参与编写,具有相当参考价值。
【新药批准和报产】
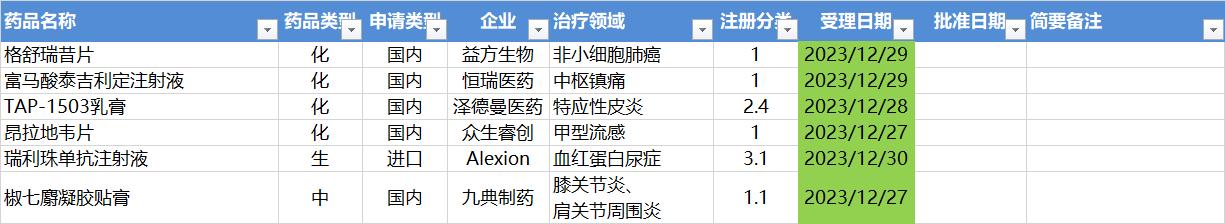
12.25-12.31,NMPA未发布新药批准,CDE受理6个NDA
注:仅列出新药(包括改良型新药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林®版权所有,未经许可不得转载
|