|
首页
>
资讯
>
欧盟新临床试验申请强制使用临床试验信息系统(CTIS)
出自识林
欧盟新临床试验申请强制使用临床试验信息系统(CTIS)
2023-02-07
从2023年1月31日起,所有在欧盟(EU)的初始临床试验申请必须通过临床试验信息系统(Clinical Trials Information System,CTIS)提交,CTIS现在是申办者和临床试验监管机构提交和评估临床试验数据的单一入口。CTIS于2022年1月31日上线,是根据欧盟临床试验法规(Regulation (EU) No 536/2014)建立的,用以支持临床试验申办者、EU成员国、EEA国家和欧盟委员会之间信息交流的在线系统。
在过去,申办者需要分别向每一个成员国药监局和伦理委员会递交临床试验申请(Clinical Trial Application,CTA),以获得批准,现在通过CTIS系统,申办者能够通过一个在线申请在多达30个欧洲国家申请临床试验许可,并允许成员国监管机构合作处理多个国家的临床试验申请、请求进一步信息、许可或拒绝临床试验以及监督已获批的临床试验。
新临床试验申请的过渡期
从CTIS上线,欧盟设置了三年的过渡期:
至2023年1月30日,申办者可以选择根据欧盟临床试验法令(Directive 2001/20/EC)或临床试验法规(Regulation (EU) No 536/2014)申请新的临床试验许可。
从2023年1月31日起,申办者必须通过CTIS提交所有新申请。
自2025年1月31日起,根据临床试验法令(Directive 2001/20/EC)批准的所有正在进行的临床试验都需要符合临床试验法规,并过渡到CTIS。
申办者手册
EMA发布的Clinical Trials Information System (CTIS) - Sponsor Handbook(CTIS申办者手册)提供了CTIS使用的关键指南、技术信息、建议、培训材料和支持性文件的汇编和参考。其主要内容包括:
- 1. 什么是CTIS及其功能
- 2. 进入CTIS–注册
- 3. CTIS中用户和组织的管理
- 4. 如何在CTIS开始临床试验申请
- 5. 从法令到临床试验法规的过渡
- 6. CTIS中的产品管理
- 7. 数据、文件和流程
- 8. 数据透明度
- 9. 安全报告义务
- 10. 支持
- 11. 其他参考文献
- 12. 首字母缩略词和术语
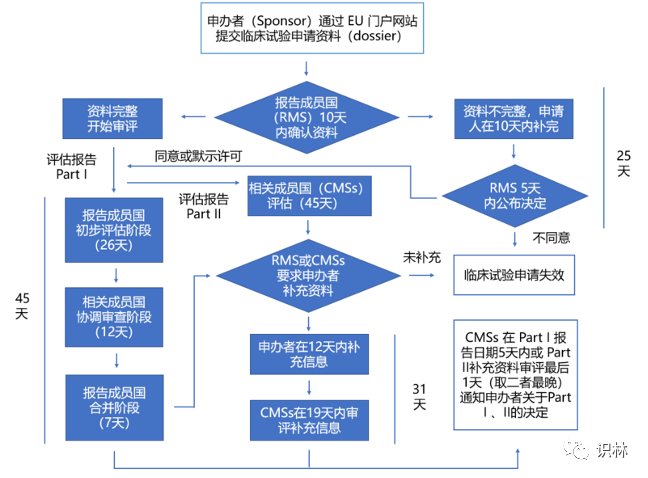
初始CTA的批准流程
参考欧盟临床试验法规(Regulation (EU) No 536/2014),初始CTA的批准流程如下:
注:Part Ⅰ包含临床试验的详细信息、申办者和产品信息,Part Ⅱ包含临床试验场地的详细信息和文档。
其他CTA评估时间线
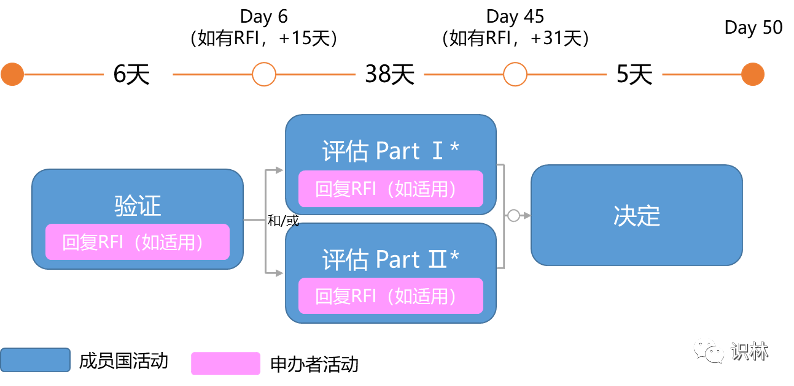
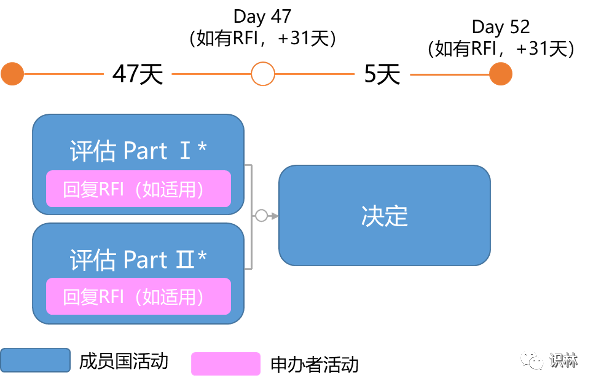
除初始CTA外,临床试验申请评估的类型还包括:实质性修改CTA(Substantial Modification CTA)、额外相关成员国CTA(Additional MSC CTA)。每个申请程序都有相应的时间表。
是指对已做出决定CTA的变更的申请。其评估时间线为:
注:回复RFI(Request for Information)仅适用于成员国提出RFI的情况。
*取决于递交的CTA,SM CTA可包含Part Ⅰ和Ⅱ,也可以仅包含Part Ⅰ或Ⅱ。
是指将之前已获批的临床试验扩展到其他成员国的申请。其评估时间线为:
*取决于递交的CTA,对于增加相关成员国的CTA,用户可能提出针对Part Ⅰ的考虑,但结论不能修改。
作者:识林-栀
识林®版权所有,未经许可不得转载
|