|
首页
>
资讯
>
欧盟新临床试验申请强制使用临床试验信息系统(CTIS)
出自识林
欧盟新临床试验申请强制使用临床试验信息系统(CTIS)
2023-02-07
从2023年1月31日起,所有在欧盟(EU)的初始临床试验申请必须通过临床试验信息系统(Clinical Trials Information System,CTIS)提交,CTIS现在是申办者和临床试验监管机构提交和评估临床试验数据的单一入口。CTIS于2022年1月31日上线,是根据欧盟临床试验法规(Regulation (EU) No 536/2014)建立的,用以支持临床试验申办者、EU成员国、EEA国家和欧盟委员会之间信息交流的在线系统。
在过去,申办者需要分别向每一个成员国药监局和伦理委员会递交临床试验申请(Clinical Trial Application,CTA),以获得批准,现在通过CTIS系统,申办者能够通过一个在线申请在多达30个欧洲国家申请临床试验许可,并允许成员国监管机构合作处理多个国家的临床试验申请、请求进一步信息、许可或拒绝临床试验以及监督已获批的临床试验。
新临床试验申请的过渡期
从CTIS上线,欧盟设置了三年的过渡期:
至2023年1月30日,申办者可以选择根据欧盟临床试验法令(Directive 2001/20/EC)或临床试验法规(Regulation (EU) No 536/2014)申请新的临床试验许可。
从2023年1月31日起,申办者必须通过CTIS提交所有新申请。
自2025年1月31日起,根据临床试验法令(Directive 2001/20/EC)批准的所有正在进行的临床试验都需要符合临床试验法规,并过渡到CTIS。
申办者手册
EMA发布的Clinical Trials Information System (CTIS) - Sponsor Handbook(CTIS申办者手册)提供了CTIS使用的关键指南、技术信息、建议、培训材料和支持性文件的汇编和参考。其主要内容包括:
- 1. 什么是CTIS及其功能
- 2. 进入CTIS–注册
- 3. CTIS中用户和组织的管理
- 4. 如何在CTIS开始临床试验申请
- 5. 从法令到临床试验法规的过渡
- 6. CTIS中的产品管理
- 7. 数据、文件和流程
- 8. 数据透明度
- 9. 安全报告义务
- 10. 支持
- 11. 其他参考文献
- 12. 首字母缩略词和术语
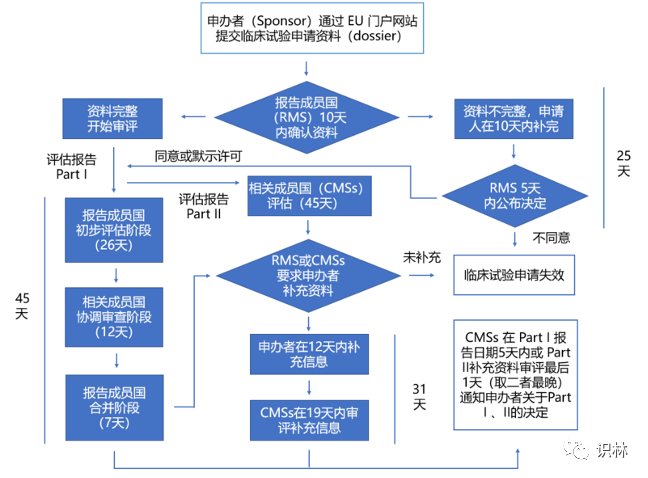
初始CTA的批准流程
参考欧盟临床试验法规(Regulation (EU) No 536/2014),初始CTA的批准流程如下:
注:Part Ⅰ包含临床试验的详细信息、申办者和产品信息,Part Ⅱ包含临床试验场地的详细信息和文档。
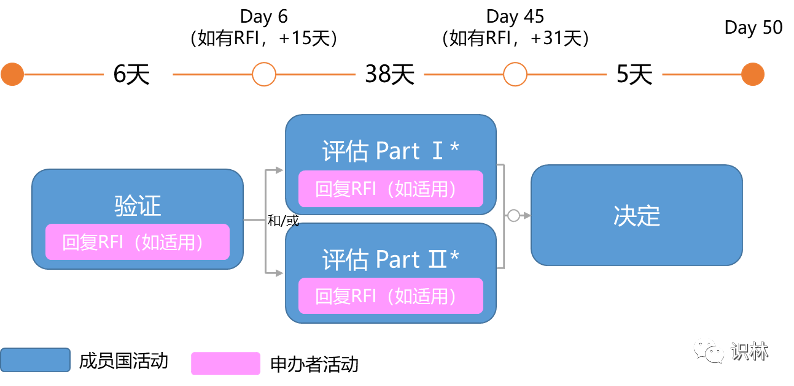
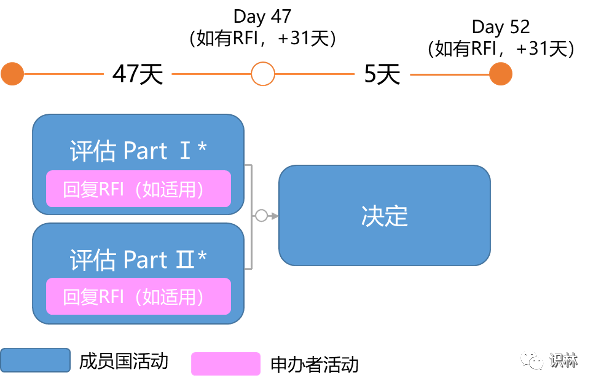
其他CTA评估时间线
除初始CTA外,临床试验申请评估的类型还包括:实质性修改CTA(Substantial Modification CTA)、额外相关成员国CTA(Additional MSC CTA)。每个申请程序都有相应的时间表。
是指对已做出决定CTA的变更的申请。其评估时间线为:
注:回复RFI(Request for Information)仅适用于成员国提出RFI的情况。
*取决于递交的CTA,SM CTA可包含Part Ⅰ和Ⅱ,也可以仅包含Part Ⅰ或Ⅱ。
是指将之前已获批的临床试验扩展到其他成员国的申请。其评估时间线为:
*取决于递交的CTA,对于增加相关成员国的CTA,用户可能提出针对Part Ⅰ的考虑,但结论不能修改。
作者:识林-栀
识林®版权所有,未经许可不得转载
适用岗位及工作建议: - 临床(Clinical):必读。确保所有临床试验遵循GCP原则,保护受试者权益,确保数据的准确性和可靠性。
- 注册(Regulatory Affairs):必读。负责确保临床试验的合规性,包括伦理审查、授权和监督。
- 质量保证(QA):必读。监督GCP的实施,确保临床试验的质量控制和质量保证。
- 药物警戒(PV):必读。负责监测和报告临床试验中的不良事件和严重不良反应。
文件适用范围:
本文适用于欧盟成员国,涉及人类使用的药品临床试验,包括化学药、生物制品和疫苗等。适用于创新药、仿制药、生物类似药和原料药等注册分类。针对Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 临床试验的GCP实施:明确所有临床试验需遵循GCP原则,保护受试者权利、安全和福祉,并确保临床试验结果的可信度。
- 受试者保护:强调在临床试验中对未成年人和无能力提供知情同意的成年人的特别保护措施。
- 伦理委员会的角色:规定伦理委员会在临床试验开始前需提供意见,并在试验过程中监督受试者保护。
- 不良事件和反应的报告:要求及时报告所有严重不良事件和严重不良反应,并在年度报告中包含所有疑似严重不良反应。
- 监管和合规性:强调成员国需采取必要措施确保临床试验的监管和合规性,包括制造、进口、标签和检查等方面。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - 临床研究部门(Clinical):需熟悉CTIS的使用流程,确保临床试验申请符合EMA要求。
- 注册部门(Regulatory Affairs):应利用CTIS进行高效的注册申报,及时更新注册信息。
- 数据管理(DM):负责确保数据的准确性和及时性,与CTIS系统同步。
文件适用范围:
本文适用于在欧盟进行的化学药、生物制品和疫苗等药品的临床试验,包括创新药和仿制药,适用于Biotech、大型药企、跨国药企等各类企业。 文件要点总结: - CTIS系统概述: 明确了CTIS系统的目的和功能,为申办者提供了一个统一的临床试验信息平台。
- 注册流程要求: 强调了通过CTIS提交临床试验申请的具体步骤和要求,包括资料的完整性和合规性。
- 数据管理与报告: 规定了申办者在CTIS中进行数据管理和定期报告的义务,确保信息的透明和及时更新。
- 监管互动: 鼓励申办者与EMA进行有效沟通,通过CTIS系统及时解决疑问和监管问题。
- 变更和更新: 明确了临床试验过程中变更和更新信息的流程,要求申办者及时在CTIS中反映这些变化。
以上仅为部分要点,请阅读原文,深入理解监管要求。 解读法规指南 适用岗位: - 临床研究部门(Clinical):必读,需根据文件要求设计和执行临床试验。
- 法规事务部门(Regulatory Affairs):必读,负责确保临床试验符合法规要求。
- 质量保证部门(QA):必读,确保临床试验的质量控制和合规性。
- 药物警戒部门(Pharmacovigilance):必读,负责临床试验期间的安全性监控和报告。
工作建议: - 临床研究部门:设计试验方案时,需确保试验的科学性和伦理性,并符合相关法规要求。
- 法规事务部门:提供法规培训,确保团队理解并能够应用法规指南。
- 质量保证部门:制定质量控制流程,确保临床试验的每个环节都达到合规标准。
- 药物警戒部门:建立监测和报告机制,确保所有不良事件得到及时处理和报告。
适用范围:
本文适用于在欧盟进行的涉及人类使用药品的临床试验,包括化学药品、生物制品等,不包括非干预性研究。适用于创新药、仿制药、生物类似药等注册分类,由欧盟发布,适用于Biotech、大型药企、跨国药企等。 要点总结: - 临床试验授权程序:强调了临床试验必须经过科学和伦理审查,并根据本法规获得授权。
- 申请提交与评估:详细规定了提交临床试验申请的流程、评估报告的内容以及决策的时限。
- 受试者保护和知情同意:确保受试者的权利、安全、尊严和福祉得到保护,并在充分知情的基础上自愿参与试验。
- 安全性报告:要求建立电子数据库用于安全性报告,并规定了不良事件和严重不良事件的报告流程。
- 临床试验的执行和监督:强调了遵守临床试验方案和良好临床实践的重要性,以及对临床试验进行适当监督的必要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |