首页
>
资讯
>
沙特 FDA 的 RAID:面向“全球新”药物的优先审评与滚动提交
出自识林
沙特 FDA 的 RAID:面向“全球新”药物的优先审评与滚动提交
2026-04-23
4月5日,沙特食品药品管理局(Saudi Food and Drug Authority,SFDA)发布了《研究与试验用药物(RAID)认定》 指南。RAID计划旨在加速未获任何其他监管机构上市许可 的创新药(“全球新”)的开发与审评。该指南将于2026年7月2日正式生效,自此SFDA加入了主流药监追求全球新药的行列。
SFDA允许申办者在临床开发的任意阶段(I期、II期或III期)提交RAID认定申请。申请时需基于已有科学或临床证据,证明该产品针对未满足的医疗需求,或相比现有治疗选项具有显著的治疗或科学价值。RAID认定的另一个前提条件是产品临床试验 在沙特阿拉伯境内开展。
申办者可在提交正式申请前申请预认定会议。预认定阶段内,申办者可针对SFDA提出的缺陷 、意见或补充信息要求修改申请。
药企获得RAID认定后的激励与权益包括:
SFDA指派一名产品经理作为专属联络点,协调RAID申请过程中的支持工作。
预认定会议邀请SFDA多学科专家组参与,提供关于RAID认定要求的初步科学和监管指导。
科学指导方面,SFDA提供科学建议和监管支持,协助申办者使开发策略符合SFDA要求。RAID认定产品可获得与SFDA及利益相关方的频繁互动机会,包括产品开发期间的多次会议。
定价规则方面,SFDA为研究性产品的定价程序提供支持,包括批准会议请求、提供定价规则咨询、与注册审评并行开展定价审评,以及启动与申办者的早期定价讨论。
监管路径指导方面,RAID认定产品在提交上市许可申请(MAA)时可获得加速评估。具体权益包括:MAA的优先审评 、滚动提交(SFDA可在申办者提交完整申请前审评部分MAA内容),以及附条件批准 (基于临床数据支持)。
根据指南附录3,申办者需按照eCTD 格式编制RAID认定申请材料。获得RAID认定后,申办者需在六个月内通过沙特临床试验登记系统提交临床试验申请 ,并同步提交产品申请。
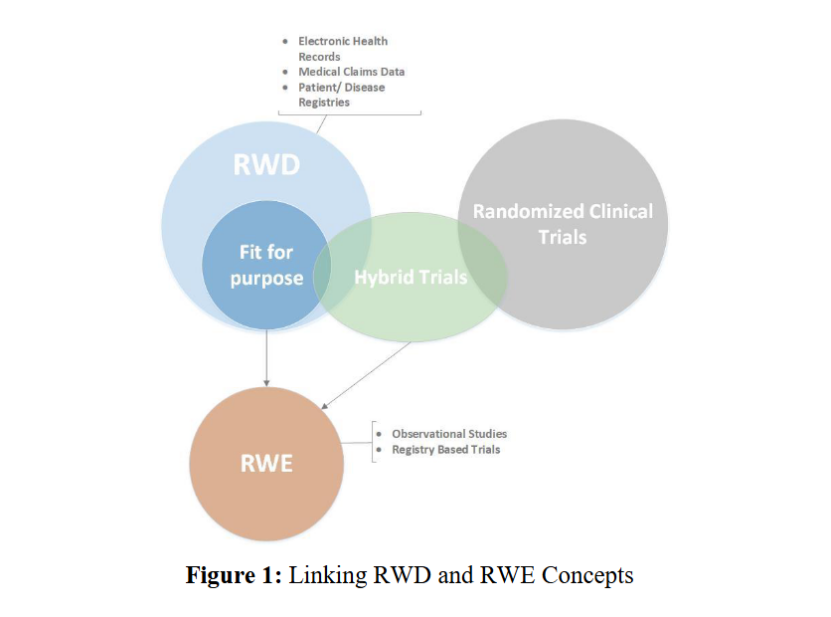
除了审评路径,SFDA的行动还涉及监管科学 前沿领域。4月20日,SFDA发布了《利用真实世界数据(RWD)和真实世界证据(RWE)支持药品上市许可有效性与安全性的框架》 。该框架提出了RWD/RWE在五类监管决策场景中的应用潜力:假设生成、单臂研究 补充、本地数据转化、先验假设支持及代表性不足的人群的研究。文件还指出需通过标准化数据模型和统计方法提升证据可靠性。与中、美、欧药监针对RWD和RWE发布的大量指南比起来,这份指南还比较浅显,但从中可以看到SFDA正在探索加入主流药监机构的行列。
SFDA的活跃背景,是沙特阿拉伯作为中东地区最大的医药市场,正处于从高度依赖进口向本土制造与生物技术创新转型的关键期。据网络数据,沙特药品市场规模已突破120亿美元,在人口老龄化、慢性病高发以及政府对生物医药产业战略支持的推动下,年增长率保持在5%左右。
作为这一转型背后的核心监管力量,SFDA自2003年成立以来,经历了从整合监管职能到引领区域标准的跨越式发展,且已加入ICH ,获WHO监管成熟度ML4评级。SFDA在中东率先推行药品追溯系统及电子提交标准,并配合国家转型计划大幅优化审批流程。作为沙特医药市场的“守门人”,SFDA也是国际药企进入中东市场的合规标杆。
作者:识林-实木
责任编辑:识林-木姜子
识林® 版权所有,未经许可不得转载。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。评估RWE替代或补充传统临床试验的可行性,制定符合SFDA要求的证据生成方案。 临床 :必读。主导RWE研究设计,确保数据采集符合“适用性”标准,采用目标试验模拟等方法控制偏倚。 数据科学 :必读。实施CDM标准化数据,开发高维倾向评分(hdPS)等分析工具处理混杂因素。 医学写作 :必读。在申报材料中明确RWE的局限性及控制措施,突出其与监管问题的关联性。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册(RA) :必读。需主导RAID申请全流程,协调eCTD资料准备,跟踪SFDA审查反馈,确保6个月内提交后续临床 trial申请(CTAp)和MAA。 临床 :必读。负责设计符合RAID要求的沙特本地临床试验方案,参与预认定会议讨论科学问题。 CMC :必读。按SFDA特定指南(如细胞治疗CMC要求)完善模块3数据,确保生产工艺描述与质量控制满足早期开发阶段标准。 以上仅为部分要点,请阅读原文,深入理解监管要求。