首页
>
资讯
>
【识林心得】“这是给我‘返回’到哪儿来了?”
出自识林
【识林心得】“这是给我‘返回’到哪儿来了?”
2026-04-18
在识林页面的众多功能按钮中,“返回”是一个看着简单却也有些用心的设计。它并非浏览器的“后退”功能,而是一条预设的知识回溯路径——点击后,即可从当前阅读的文档,直接跳转至其所属的官方通告、资源索引、历史版本或案例合集页面。
本文通过五个典型场景,展示这一小按钮如何帮助识林用户快速定位文件来源、查阅配套资料、追溯法规沿革,让每一次“返回”都变得更有价值。
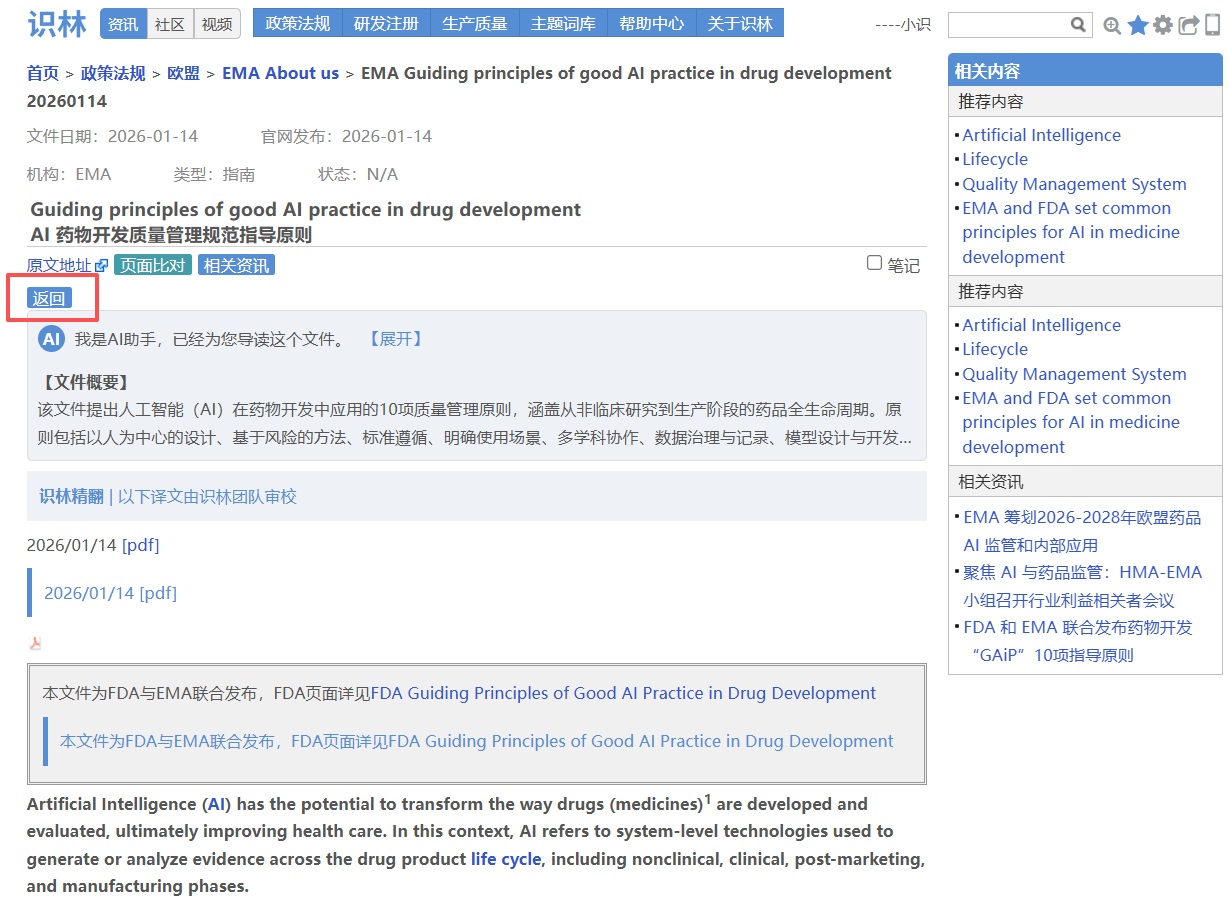
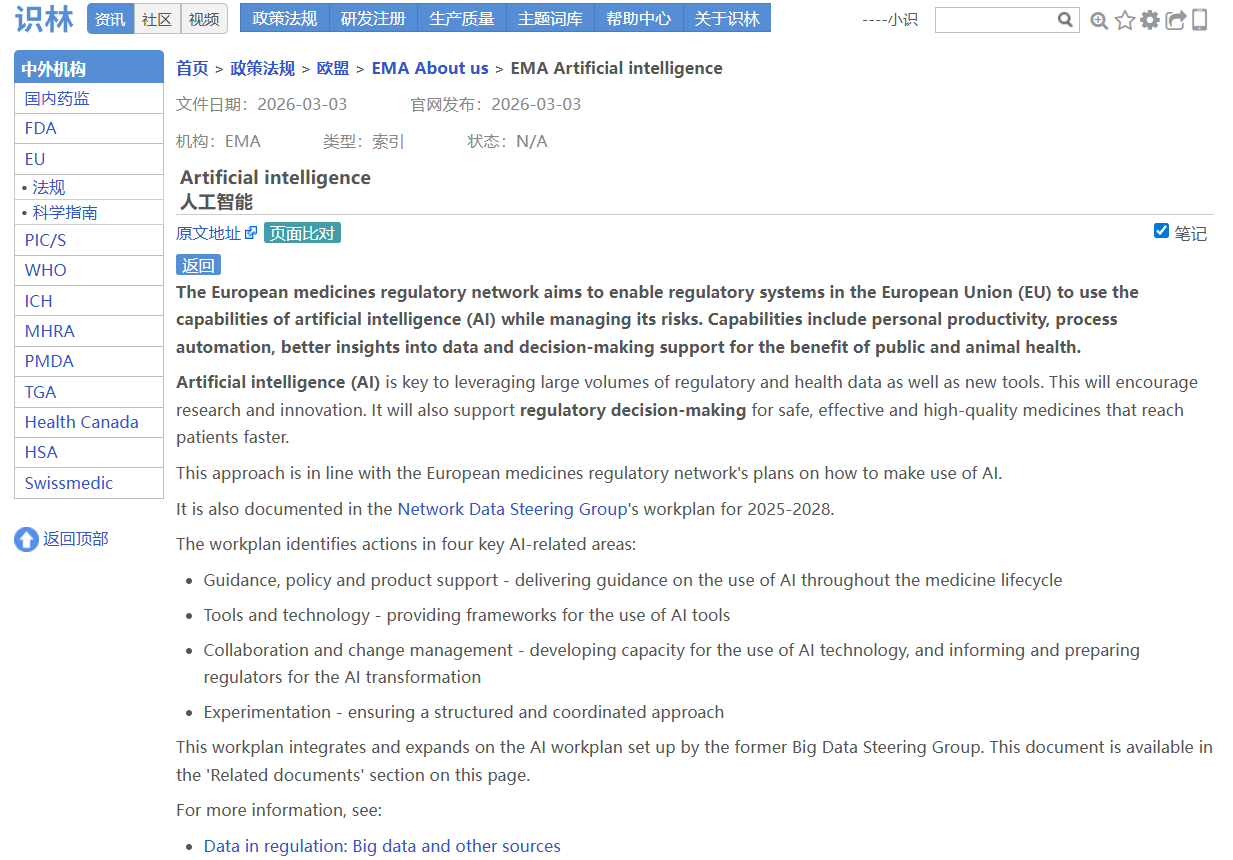
场景1:从欧美GAiP【返回】 EMA的AI索引,一览EMA的AI布局
AI 在药物开发和监管中的应用日益增多,但相关指南分散在不同页面,官方来源也需要反复核对。识林将《AI药物开发质量管理规范指导原则》 与EMA人工智能总索引页 通过“返回”键关联,从一份文件即可回溯至EMA全部AI资源库(思考性文件、AI观察站报告、Scientific Explorer工具及首个AI临床工具资质 认证案例),并可通过“原文地址”进一步溯源至EMA官网原始发布页面,可供快速定位、交叉核对,确保每一份文件都有据可查。
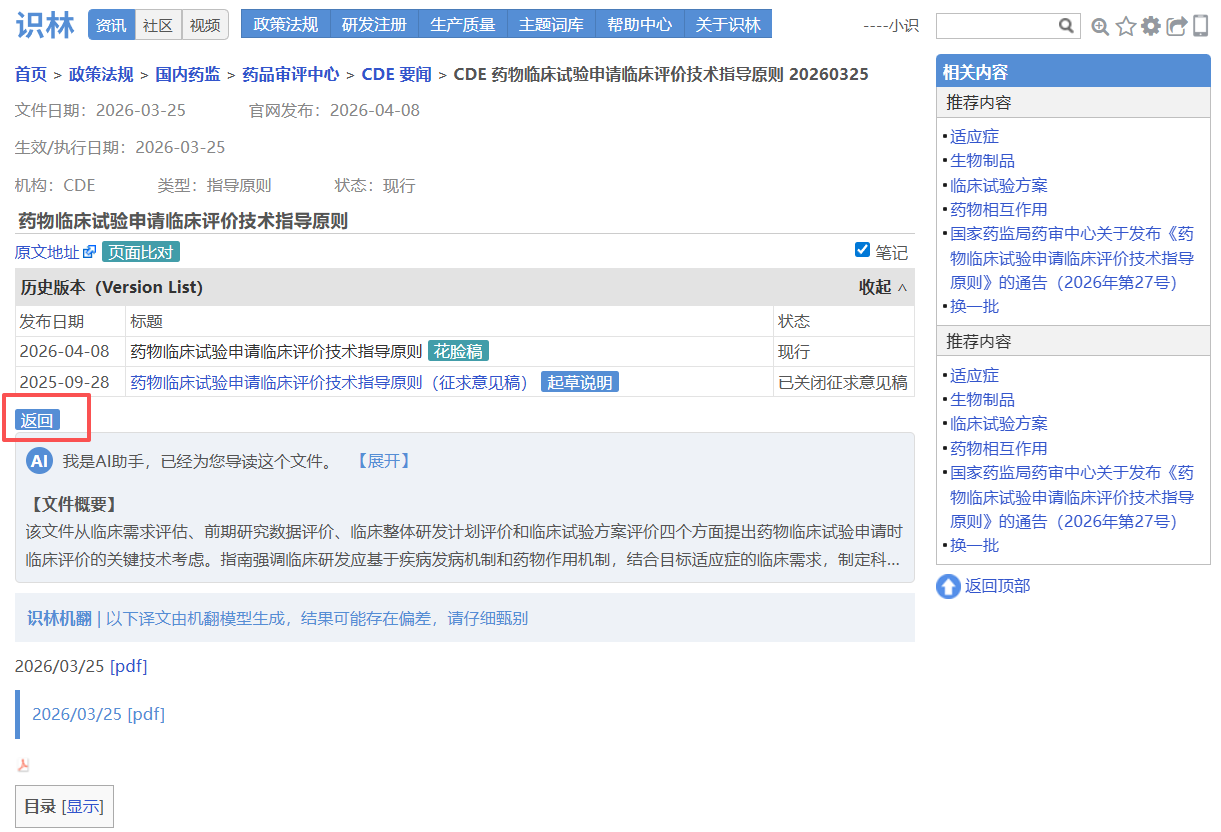
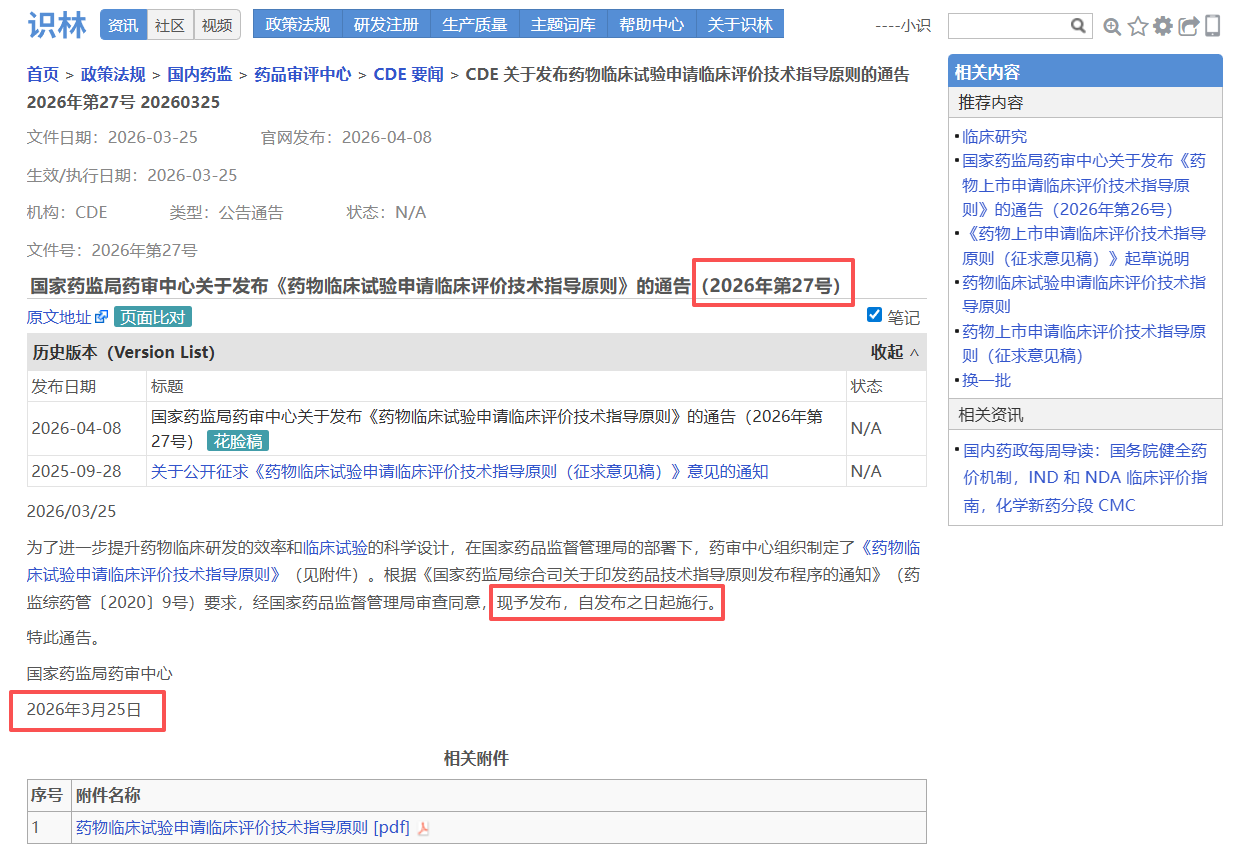
——从—— ——返回到—— 场景2:从CDE指南正文【返回】 发布通告,查询实施日期和文件号
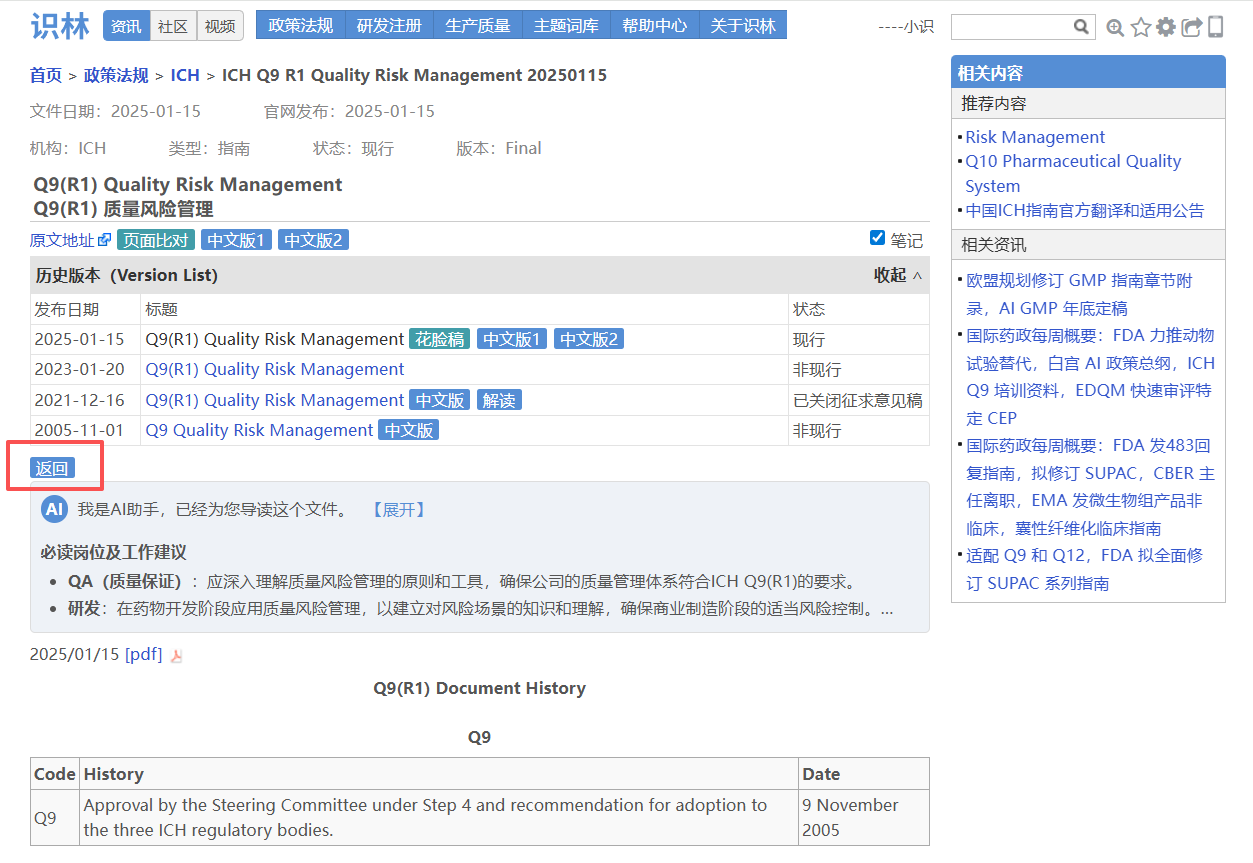
大家熟悉的CDE指导原则,其技术要求集中在指导原则正文,但官方发布背景、文件号、生效日期均在另一份通告中。例如从《药物临床试验申请临床评价技术指导原则》 正文点击“返回”即可直达通告页面 ,确认文件编号(2026年第27号)、发布日期和施行日期,并可下载原始PDF附件。对于需要确保申报合规的注册和临床研发人员,这一设计让技术内容与官方依据始终对应,避免因信息分离导致疏漏。另外,这也是考虑到用户们常用“27号文”来指代指导原则的实际场景。
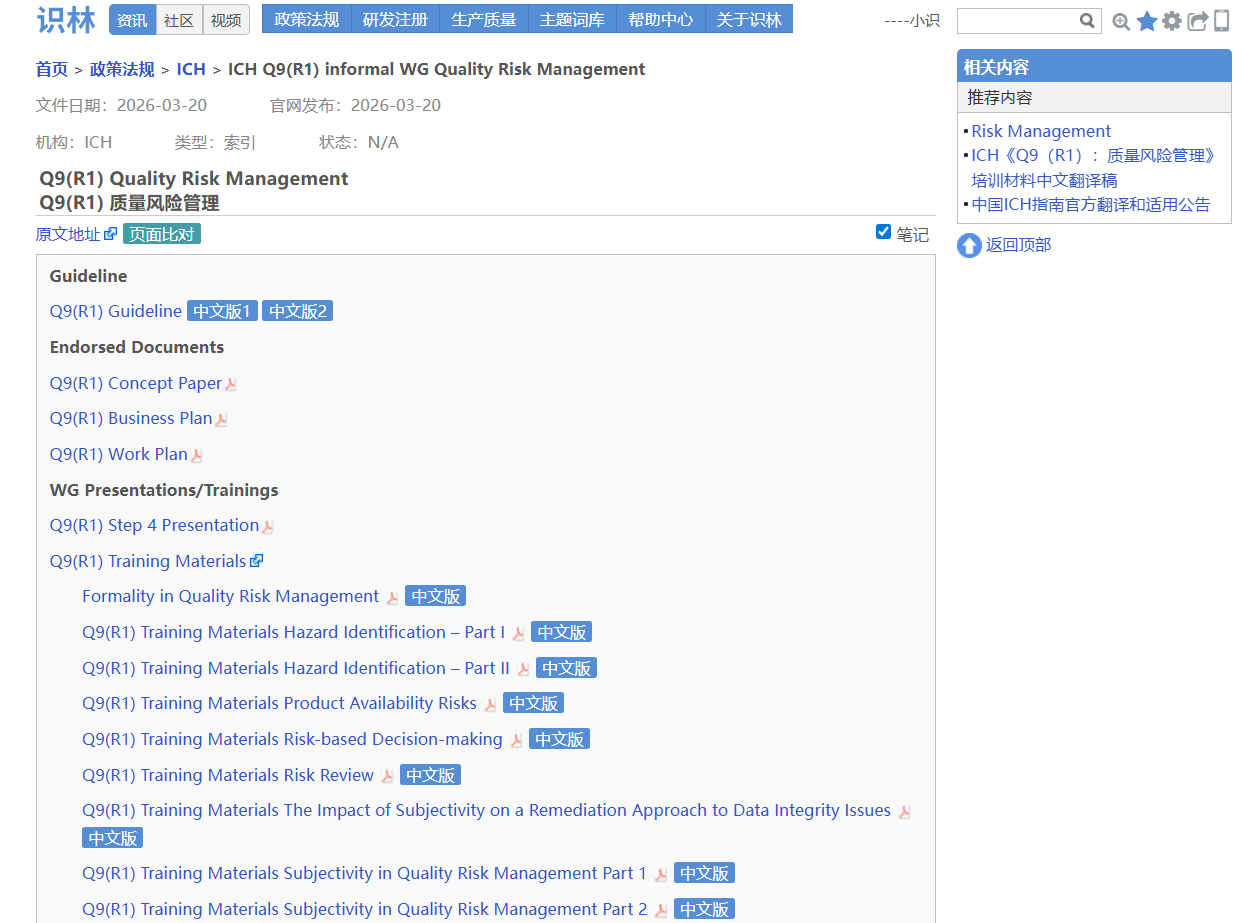
——从—— ——返回到—— 场景3:从ICH Q9【返回】 查阅历史版本,中文版以及最新培训资料包
ICH 《Q9(R1)质量风险管理》指南 配套的培训材料、概念性文件等分散在不同页面,且多为英文原文。当您找到Q9页面,点击“返回”即可打开完整索引 ,查看完整文件清单,包括CDE 和CFDI 的两份中文翻译版,以及最近更新的培训资料包。ICH 的官方PPT完整细致,具有相当的学习参考价值。
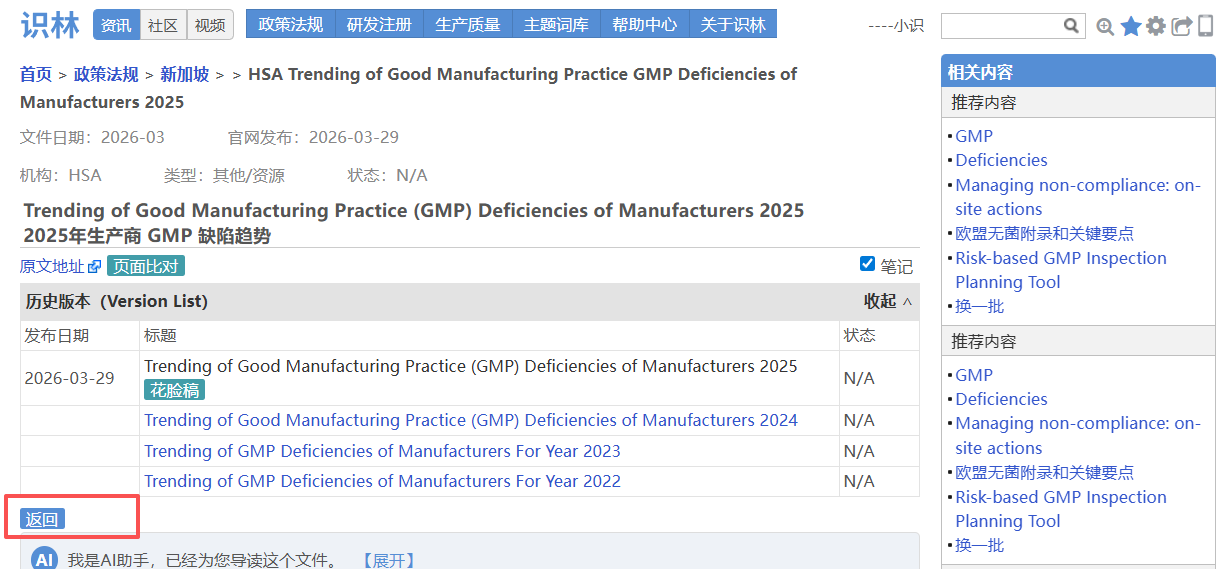
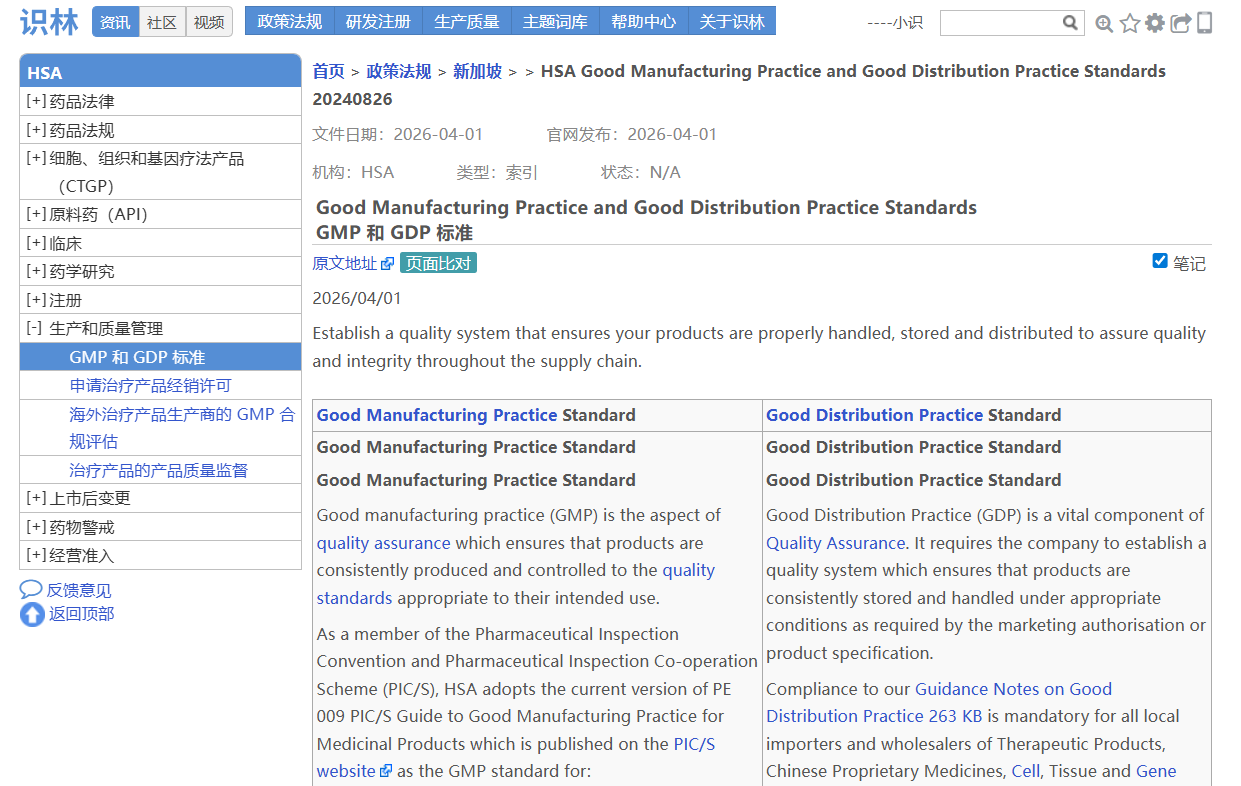
——从—— ——返回到—— 场景4:从新加坡GMP检查总结【返回】 查询新加坡GMP法规指南
新加坡卫生科学局(HSA)作为PIC/S正式成员,其GMP 标准、GDP 要求、历年缺陷 趋势及配套指南是企业现场检查准备的参考依据之一。当用户找到《2025年生产商GMP缺陷趋势》 (例如从识林发布的专题文章《新加坡HSA总结2025年药品GMP检查缺陷》 找到),此时点击“返回”即可抵达HSA的“GMP和GDP标准 ”总览页面,回溯至HSA全部资源库,涵盖GMP(PIC/S PE 009-17及附录)、GDP、历年GMP/GDP缺陷趋势分析(2023-2025)、场地主文件编制指南、基于健康暴露限值的交叉污染 控制指南等关键文件。
——从—— ——返回到—— 场景5:从一封日本PMDA“橙色警告信”【返回】 完整清单
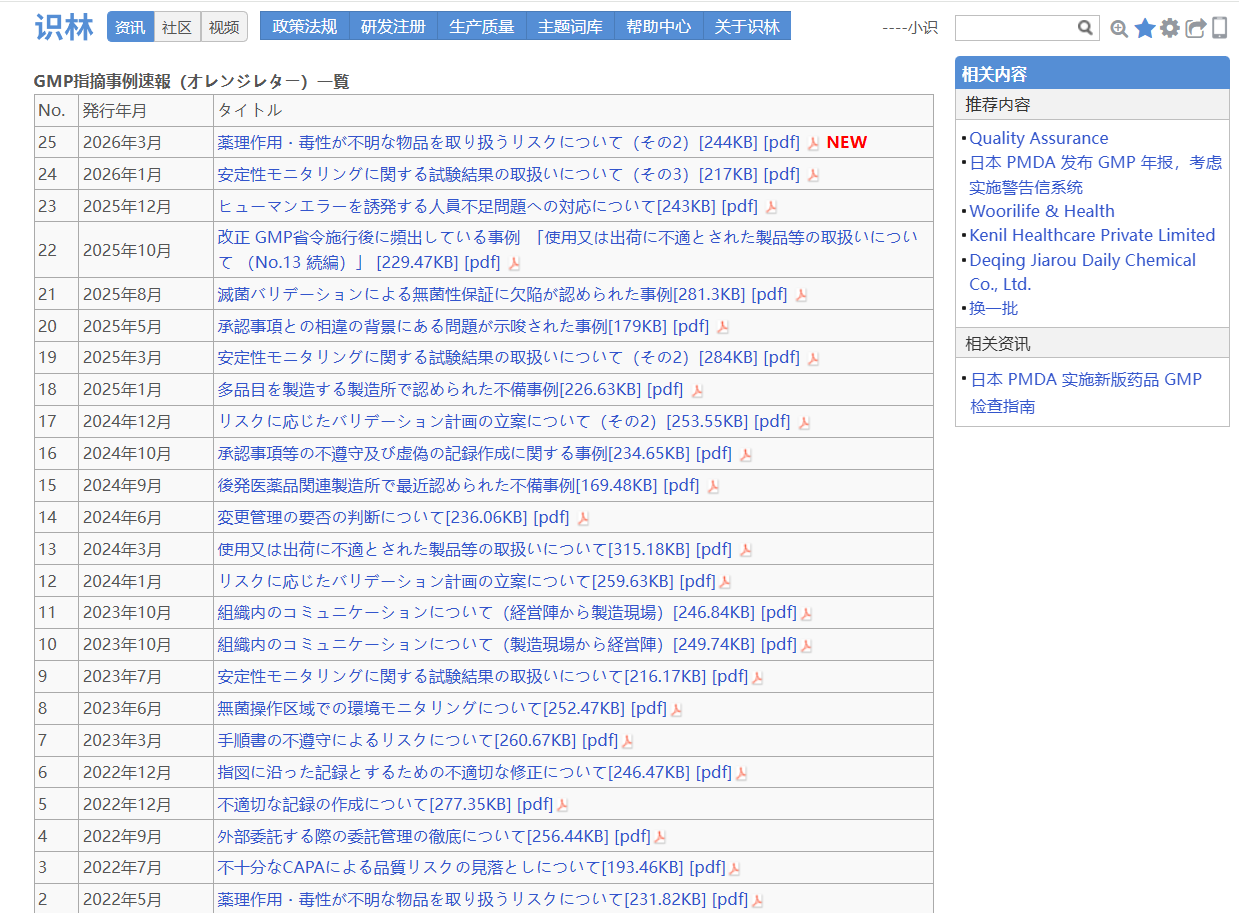
日本药品和医疗器械管理局(PMDA)“橙色警告信”是PMDA分享的GMP检查缺陷典型案例。识林将单份警告信 与《品質確保に関する取り組み》 页面通过“返回”键关联,从一份案例即可回溯至全部橙色警告信列表、GMP/GCTP年度报告等资源。PMDA发布的GMP/GCTP年度报告详细介绍了其在药品质量管理方面的业务实施与未来展望;另外,页面列出的GMP橙色警告信索引涵盖稳定性 试验结果处理、无菌 性保证缺陷等具体问题案例,为行业提供了实用参考。
——从—— ——返回到—— 更多识林心得,请见【帮助中心】 。
责任编辑:识林-木姜子
识林® 版权所有,未经许可不得转载。
岗位必读建议:
QA:应深入理解质量风险管理流程,确保生产和质量控制符合要求。 注册:需熟悉质量风险管理在药品注册中的应用,确保注册文件的合规性。 研发:应将质量风险管理融入新药开发过程,以降低后期风险。 适用范围说明:
文件要点总结:
风险管理流程 :强调建立全面的风险管理流程,包括风险识别、评估、控制和沟通。风险评估方法 :明确了风险评估的方法和工具,如FMEA(失效模式与影响分析)。风险控制措施 :规定了风险控制措施的选择和实施,以降低药品生产过程中的风险。质量体系整合 :鼓励将质量风险管理整合到企业的质量管理体系中,以提高整体质量管理水平。持续改进 :强调质量风险管理是一个持续的过程,需要定期回顾和改进。以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
临床(必读) :需依据指南优化试验设计,重点评估剂量递增、安全性监测和终点选择;建议建立跨学科团队审核方案合规性。 注册(必读) :应结合指南要求完善临床试验申请资料,确保非临床与临床数据逻辑连贯;需跟踪SUSAR报告提交的时效性。 研发(必读) :在早期研究中需关注种族敏感性、特殊人群数据缺口,为后期临床方案提供支持;建议提前规划儿科研究计划(PSP)。 以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):必须理解并实施质量风险管理流程,确保所有质量风险管理活动与风险水平相适应。 研发:在产品设计和开发阶段应用质量风险管理,以识别潜在风险并采取预防措施。 生产运营:在生产过程中应用质量风险管理,以控制和降低风险,确保产品质量。 注册:在药品注册申报过程中,使用质量风险管理结果支持监管要求的满足。 市场:了解质量风险管理对市场策略的影响,特别是在产品可及性风险管理方面。 文件适用范围:
文件要点总结:
质量风险管理原则 :强调基于科学知识评价质量风险,保护患者,且管理过程应与风险水平相适应。基本流程 :明确了质量风险管理的基本流程,包括职责、启动、评估、控制、沟通和回顾。风险管理方法学 :介绍了质量风险管理的正式性、基于风险的决策和降低主观性的方法。应用范围 :讨论了质量风险管理在制药行业和监管机构中的应用,包括对产品可及性风险的管理。工具与方法 :提供了多种质量风险管理工具和方法,如FMEA、FTA、HACCP等,并讨论了它们的潜在应用。以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: 注册 :评估AI应用是否符合GxP及区域性法规,更新申报材料模板。 研发 :在模型设计中嵌入可解释性验证,建立跨学科协作机制。 QA :制定AI生命周期监控计划,确保数据治理符合GxP追溯要求。 临床 :验证AI辅助试验设计的性能评估方法,明确使用场景边界。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: QA :优先审核厂房设备验证、环境监测程序及文件控制流程,建立缺陷案例库用于内部培训。 生产 :针对无菌操作、物料标识和清洁验证等高频问题修订SOP,强化现场操作合规性审计。 注册 :关注缺陷数据对上市后变更申报的影响,预判监管问询焦点。 物料管理 :优化温控系统校准和物料追溯记录,确保关键数据实时录入。 以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议 QA(质量保证) :应深入理解质量风险管理的原则和工具,确保公司的质量管理体系符合ICH Q9(R1)的要求。研发 :在药物开发阶段应用质量风险管理,以建立对风险场景的知识和理解,确保商业制造阶段的适当风险控制。生产 :在生产过程中实施质量风险管理,以识别和控制潜在的质量风险,确保产品质量和供应的连续性。注册 :在药品注册过程中,利用质量风险管理数据和分析来支持注册申请,展示产品的风险控制措施。文件适用范围 本文适用于化学药、生物制品和生物技术产品的质量风险管理,包括原料药、创新药、仿制药和生物类似药。适用于全球范围内的Biotech、大型药企、跨国药企、CRO和CDMO等不同企业类别,由ICH发布。
文件要点总结 质量风险管理定义 :“质量风险管理是一个系统的过程,用于评估、控制、沟通和回顾药品质量的风险。”风险管理方法 :介绍了多种风险管理工具,如FMEA、FMECA、FTA、HACCP等,以及它们在药品质量风险管理中的应用。风险管理过程 :强调了风险评估、风险控制、风险沟通和风险回顾的重要性,并提供了相应的步骤和方法。风险管理的整合 :讨论了如何将质量风险管理整合到行业和监管操作中,以及如何通过培训和教育提高行业和监管人员对质量风险管理的理解和信心。产品可用性风险 :特别强调了质量/生产问题对产品供应的影响,以及如何通过质量风险管理来预防和减轻由此产生的风险。以上仅为部分要点,请阅读原文,深入理解监管要求。