首页
>
资讯
>
创新药的中美双报
出自识林
创新药的中美双报
2026-01-19
一、背景
中美两国在药品注册 分类和监管体系上存在差异。FDA通常将在美国首次批准的新分子实体 视作“创新药”统计。而中国在2016年监管改革后建立了新的药物注册分类 ,强调对“全球首创新药”与“已在境外上市的进口新药”进行区分。创新化药和创新生物制品的注册分类均为1,而境外已上市化学药和境外已上市生物制品 分别对应 5.1 类和3.1类。对进口创新药的特殊管理,在制度设计上以缩短创新药中国上市滞后为核心目标,通过明确境外原研药的注册路径,鼓励全球创新药在研发和申报阶段即纳入中国,实现更高程度的同步上市。
在改革初期及更早阶段,监管实践中通常要求境外已获批药品在中国开展桥接试验,以验证 其在中国人群中的安全性和有效性,导致进口创新药在中国上市普遍存在明显滞后。随着中国正式加入 ICH (International Council for Harmonisation,国际人用药品注册技术协调会),并逐步与 ICH 技术指南接轨,中国监管部门显著提升了对境外临床试验 数据的科学认可度。在此基础上,监管政策进一步鼓励国际多中心临床试验 (international multi-regional clinical trials, MRCTs)在早期即纳入中国研究中心和中国受试者 ,使中国人群数据成为全球关键注册证据的一部分。
在政策引导下,显著缩短了创新药在中国上市的时间差,使部分创新药得以实现中美同步获批,或至少大幅降低中国相对于美国的上市滞后程度。
二、中美双报概况(2015–2025)
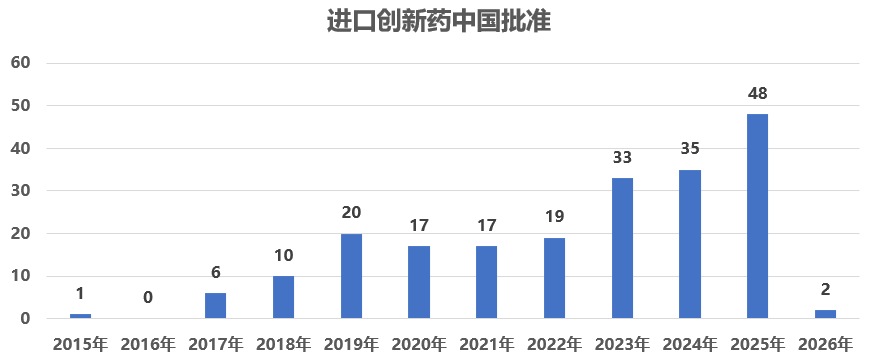
2015年至2025年间,美国FDA的CDER共批准了511个新分子实体创新药[1] ,其中231个(45%)已经在中国获得上市批准。这些进入中国的创新药大多数为进口原研药:据统计,在这231个中国上市的品种中[2] ,202个是由跨国药企开发的进口创新药,11个是中国本土研发的国产创新药,4个是化药国内仿制进口原研[3] ,2个是国内改良进口原研[4] ;另有12个品种由于在中国上市已久无法明确判定首次获批产品来源。图 1反映出过去十年里,中国市场对美国创新药的引进速度逐步提升。
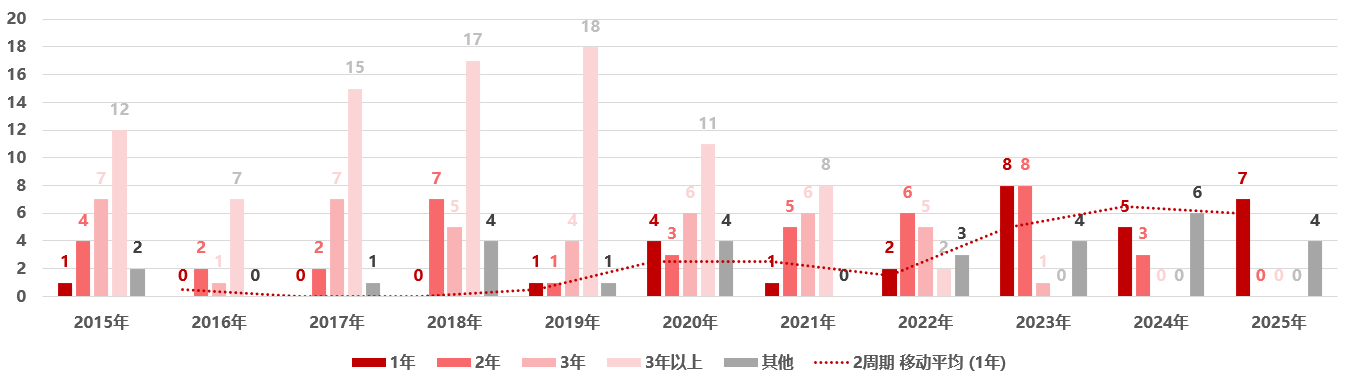
图 2显示,中美创新药同步获批(批准时间相差在1年以内)的情况呈上升趋势。例如,2015 年美国批准的新药中仅有 1 个在一年内引入中国,而到 2023 年这一数字增至 8 个,反映出近年来中美创新药上市时间差整体有所缩小。2025 年,美国当年批准的新药截至 2026 年初已有 11 个获中国批准上市,其中 8 个为进口创新药(阿夫凯泰在中国的批准时间早于美国),另有 3 个为中国本土研发的新药。这一趋势并非单纯由审评时效变化所驱动,而是在监管环境持续优化的背景下,审评审批优化和企业全球同步研发与申报策略调整的综合结果,显示部分创新药正逐步实现中美市场的准同步进入。
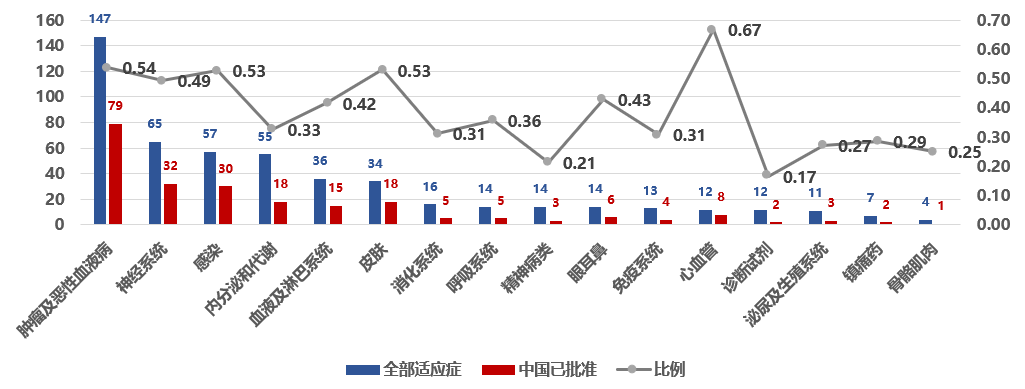
不同治疗领域中,美国产新药进入中国的比例存在差异。
如图 3所示,在美国创新药较为集中的领域,中美双报比例相对更高,50%左右。例如肿瘤及恶性血液病领域,美国2015–2025年共批准新药147个,其中有79个已进入中国(54%)。同样,感染领域有53%的美国新药已在中国上市,神经系统领域49%。这意味着在抗癌药、抗感染药等重大领域,进口创新药更频繁地被纳入中国市场。相比之下,内分泌代谢等领域比例偏低(33%)。部分领域呈现出一定特殊性,例如心血管领域虽然总体新药数量有限,但中美双报比例较高(67%)。总体来看,过去十年中,越来越多进口创新药通过同步或近同期开发与注册路径进入中国市场,在重大疾病领域,中美创新药在两国市场的上市覆盖呈现出更高的一致性。
三、进口创新药在中国的上市路径分析
注册分类
表格 1 进口创新药注册分类(2015至今批准的进口创新药)
注册分类
进口创新药
说明
化药 1
25
境内外均未上市的创新药:指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。
化药 2.45
6
境内外均未上市的改良型新药 :含有已知活性成份的新适应症 的药品。
化药 36
4
境内申请人 仿制境外上市但境内未上市原研药品的药品。该类药品应与参比制剂 的质量和疗效一致。
旧化药3.17
1
已在国外上市销售的制剂及其原料药 ,和/或改变该制剂的剂型 ,但不改变给药途径 的制剂
化药 5.1
98
境外上市的药品申请在境内上市:境外上市的原研药品和改良型药品申请在境内上市。改良型药品应具有明显临床优势。
生物制品 1
9
创新型生物制品:境内外均未上市的治疗用生物制品。
生物制品 2.18
1
改良型生物制品:在已上市制品基础上,对其剂型、给药途径等进行优化,且具有明显临床优势的生物制品。
生物制品 2.29
4
改良型生物制品:增加境内外均未获批的新适应症和/或改变用药人群。
生物制品 3.1
34
境内或境外已上市生物制品:境外生产的境外已上市、境内未上市的生物制品申报上市。
/
26
未表明注册分类25;1个未查到受理号
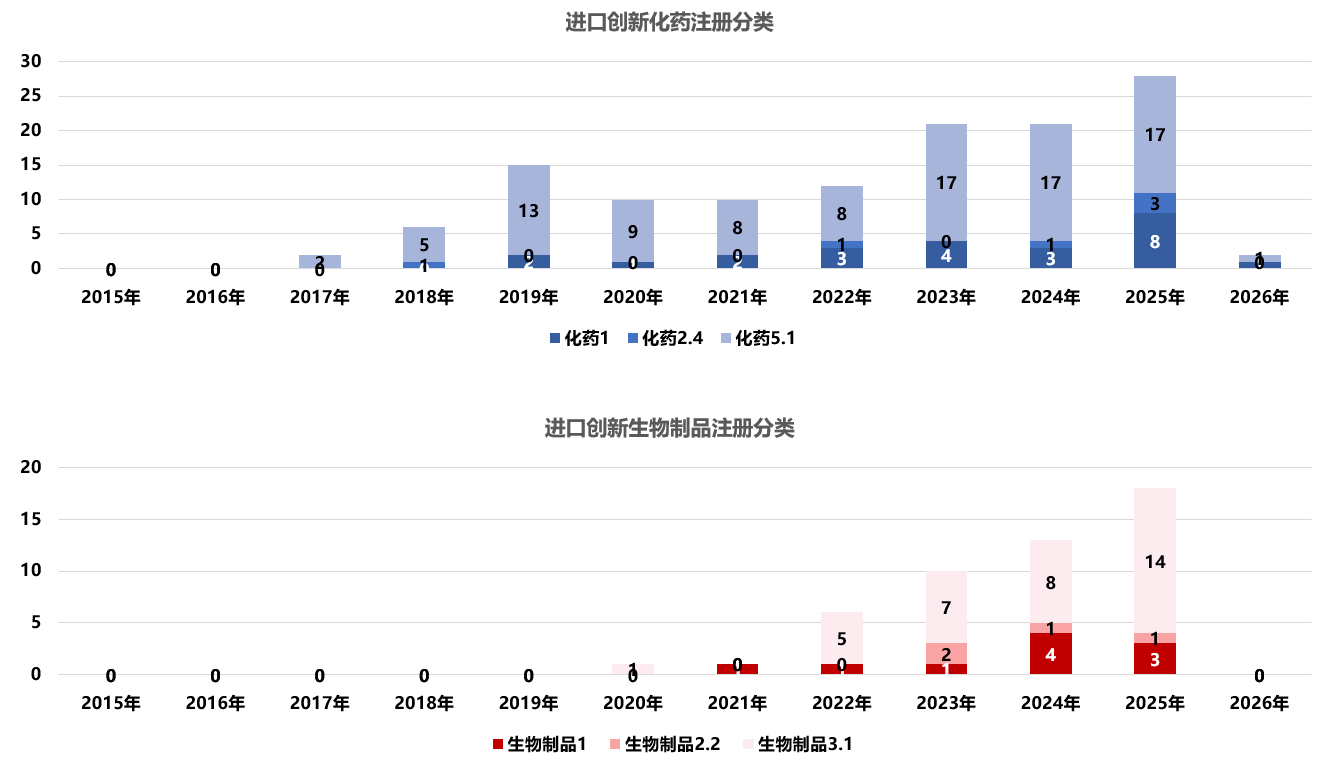
2015年至今[10] ,中国共批准了202个由跨国药企开发的进口创新药。表格 1和图 4显示,大部分在中国申报时采用了“境外已上市药品境内上市”的注册路径,即化学药5.1类或生物制品3.1类。5.1类指的是已在境外上市的原研药或改良型药品申请在中国上市(要求改良型药品具有明确临床优势);3.1类则是针对治疗用生物制品,指境外生产的境外已上市、境内未上市的生物制品申报上市。这一注册分类结构表明:在过去十年中,引入中国的创新药以海外原研药为主。另外,图 4显示,无论是化药还是生物制品 ,境内外同步研发的创新药(1类)正在逐步增加。
临床策略
2017年中国加入ICH后,监管机构加速与国际接轨,逐步完善境外临床试验数据接受政策。国家药品监督管理局(NMPA)于2017年底启动进口药品审评改革,2018年7月发布了《关于接受境外临床试验数据的技术指导原则》 ,明确了接受境外临床数据 的原则和要求。该指导原则强调申请人在提交海外试验数据时需确保数据质量符合国际公认的GCP 标准,并评估中国人群与海外受试人群之间的潜在种族差异。同时,NMPA鼓励申办者通过国际多中心临床试验(MRCT)在新药全球研发早期即同步纳入中国受试者,以避免不必要的重复研究和加速中国患者受益。这一系列政策措施(合称“接受境外临床数据政策”)极大加快了海外创新药进入中国的进程。
需要澄清的是,进口创新药在中国注册并非只能在“国际多中心试验”和“国内桥接试验”之间做出二选一的策略选择。实际情况下,同一药物既可在全球关键试验中纳入中国患者,又根据需要在中国额外开展桥接研究 。例如,一些新药在全球Ⅲ期试验包括中国受试者的同时,仍另行进行了中国受试者参与的药代动力学 桥接试验,以补充种族差异数据。反之,对于未在全球试验中纳入中国人群但符合一定条件的药物,监管部门也可能豁免在中国试验要求,在无中国受试者数据的基础上批准其上市。根据NMPA发布的技术要求,如果全面评估证明境外数据对中国患者的有效性和安全性具有充分支持且不存在显著种族差异,
可考虑减少或免除在中国开展临床试验;只有在缺乏中国人群数据或发现潜在种族差异时,才需要考虑在国内开展桥接试验以补充验证。
表格 2 进口创新药国际多中心临床试验和国内桥接试验开展情况[11]
国际临床试验
总数
无中国人
有中国人
125
76
49
II 期
29
20
9
III 期
96
56
40
国内桥接
无
57
31
26
I 期
40
20
20
II 期
15
15
0
III 期
13
10
3
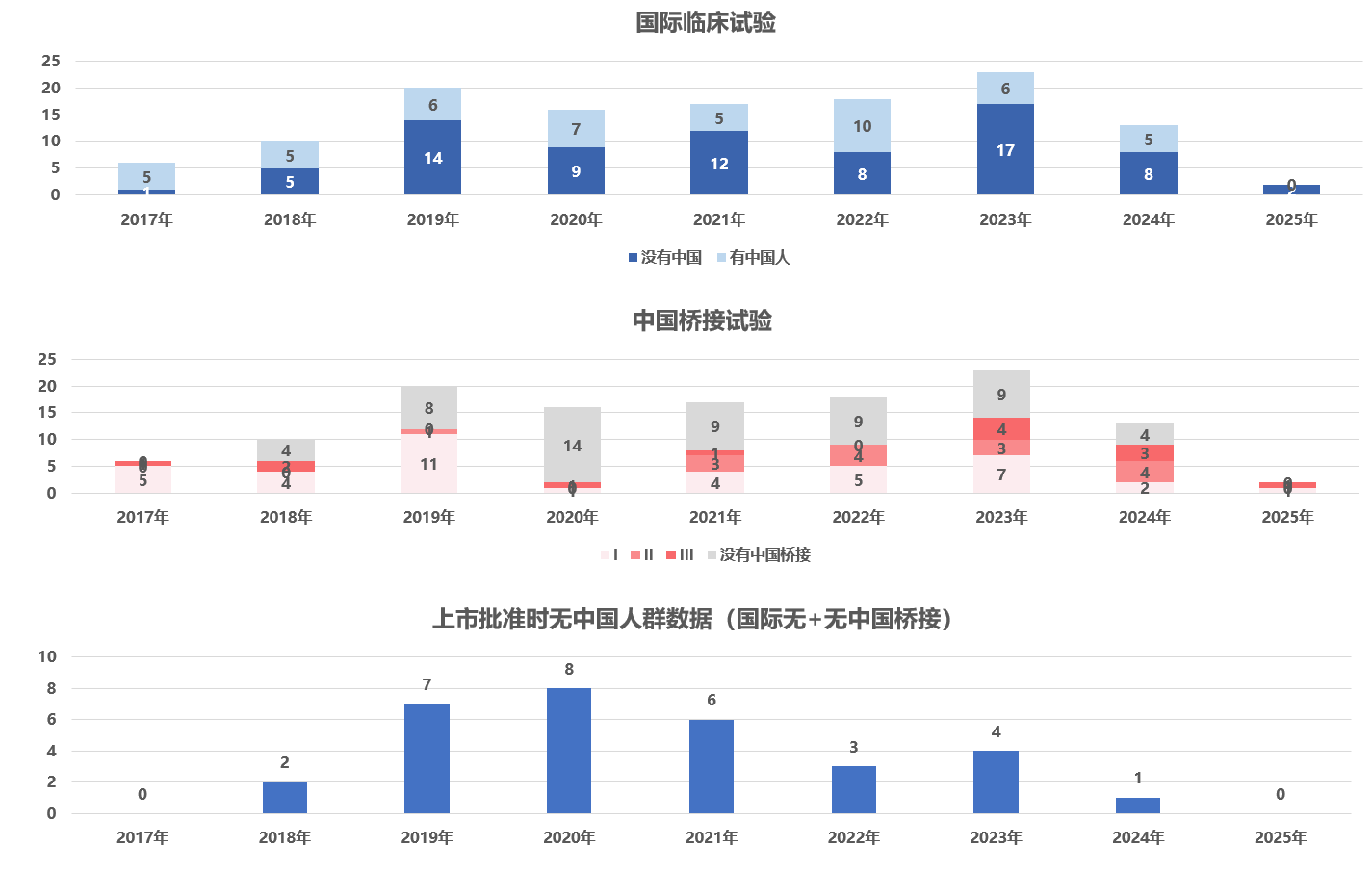
基于国家药监局药品审评中心公开的审评报告资料,我们对2015至今获批的进口创新药进行了统计分析(涵盖125个进口创新药)。表格 2显示,其中76个(61%)的药物在其全球关键临床试验中纳入了中国受试者;同时有68个(54%)药物在中国开展了某种形式的桥接试验(包括药代动力学 试验或关键性补充疗效试验)。值得注意的是,这两类并非互斥:相当一部分新药既有中国受试者参与的国际多中心临床数据,又额外进行了中国桥接研究以加强本土数据支持。与此同时,31个药物(25%)在获批时无中国人群临床试验数据——既未包含中国受试者,也未进行国内桥接试验。中国参与的国际多中心和国内桥接试验这两种路径并存互补,为海外新药进入中国提供了多元化的临床数据支持策略。
表格 3显示,不同治疗领域的新药在中国患者参与全球试验和桥接试验方面存在差异。[12] 以肿瘤及恶性血液病领域为例,46%创新药在国际多中心关键试验中未纳入中国受试者,而主要通过在国内开展桥接试验来补充本土数据后获批。这一模式在感染疾病领域表现相似(48%)。
相比之下,内分泌与代谢领域的新药在国际临床开发中更早期地纳入了中国患者。在该领域,多数进口新药在全球关键试验阶段已包含中国受试者。然而,来自国家药品监督管理局药品审评中心(CDE)公开资料的更新数据显示,在这类11个新药中虽有7个在国际多中心试验中纳入了中国受试者,但其中仍有4个在中国开展了桥接试验。这表明即使国际多中心试验已包含中国人群数据,上市前在中国额外进行桥接试验的做法仍然常见。需要强调的是,上述数据来源于CDE公开资料,研究结论受限于样本量 ,因而具有一定局限性。
已有公开数据显示,中国患者参与国际多中心试验的程度在不同治疗领域之间存在差异。在肿瘤和感染等重大疾病领域,相当比例的进口新药在其全球关键临床试验中未纳入中国受试者,通常通过在国内开展桥接试验的方式补充本土数据支持后获得批准。相比之下,内分泌和代谢疾病领域更多新药在国际多中心试验纳入了中国患者参与;但即便如此,公开数据表明,这些药物中多数仍在国内开展了桥接试验,提示国际多中心纳入中国数据并不必然意味着桥接试验的豁免。其他领域如神经系统、心血管及免疫系统用药等,其中国参与情况和桥接策略的分布更为多样,尚难以归纳为单一趋势。
上述差异可能反映了多个因素的综合作用,包括各适应症领域对种族差异敏感性的不同、临床替代治疗选择的可及性、以及海外研发进程中中国研究中心的介入时机等。此外,近年来随着中国监管政策逐步与国际接轨,申办方在全球同步开发阶段更倾向于主动纳入中国研究中心,也可能促使国际多中心数据中中国人群比例上升。鉴于本研究仅基于CDE公开审评资料中披露的125个新药案例,样本覆盖范围有限,所观察到的趋势尚不足以全面代表全部获批进口药物,相关结论仍需在更广泛的数据基础上进一步验证。
表格 3 不同适应症领域的国际多中心中国人涵盖以及中国桥接研究开展情况
国际多中心是否包含中国人
总数
无中国人
有中国人
是否有中国桥接研究
有
无
有
无
适应症领域
肿瘤及恶性血液病
50
23
6
6
15
感染
23
11
7
4
1
内分泌和代谢
11
2
2
4
3
皮肤
11
3
5
3
0
神经系统
8
1
5
0
2
血液及淋巴系统
9
3
2
2
2
心血管
4
1
1
2
0
免疫系统
4
1
1
2
0
眼耳鼻
2
0
1
0
1
呼吸系统
2
0
1
0
1
泌尿及生殖系统
1
0
0
0
1
总和
125
45
31
23
26
上市后要求
在审评过程中,部分进口创新药虽获得批准,但监管部门仍附加了不同形式的上市后临床相关要求。表格 4显示,在125个样本药物中,有58个(46%)未被提出任何临床相关要求;39个产品(31%)需“完善临床数据”,通常是补充长期随访、真实世界 表现或海外既有承诺性研究;另有28个(22%)被明确要求“开展相关研究”,即在中国本土进一步生成临床证据。
数据显示,上市后要求的提出与是否具备中国人群临床数据 密切相关。对于全球临床试验 未纳入中国、且审批前亦未进行桥接研究的药物中,55%被要求在中国开展补充性研究。在国际多中心未涵盖中国但已开展桥接的情形下,16%被要求在中国开展补充性研究。而在中国受试者已参与国际试验的情况下,无论是否有中国桥接研究,额外在中国开展补充性研究的可能性下降到8%~9%。
综上可以看出,中国监管机构在进口创新药审评中,对上市后临床要求的设置具有分层特征,其核心考量与中国人群临床证据的充分性高度相关。对于中国受试者参与程度较低或缺失的药物,监管部门更倾向于通过上市后研究或数据补充来弥补不确定性;而当国际多中心研究已覆盖中国人群时,进一步提出本土补充研究的比例显著降低。这表明,上市后要求并非普遍性或惩罚性措施,而是作为审评决策的一种风险调节工具,用于在加快药物可及性的同时,逐步完善中国人群相关证据。需要说明的是,本分析基于公开审评资料与有限样本量 所得,相关结论主要反映总体趋势,仍有待在更大样本和更长时间维度下进一步验证 。
表格 4 进口创新药的上市后要求情况
国际多中心是否包含中国人
总数
无中国人
有中国人
是否有中国桥接研究
有
无
有
无
上市后要求13
无临床相关要求
58
18
10
17
13
完善临床数据
39
20
4
4
11
开展相关研究
28
7
17
2
2
总和
125
45
31
23
26
四、国产创新药在美国的上市路径分析
过去十年,随着中国新药研发实力的提高,已有多款国产创新药成功登陆美国市场。根据统计,截至2025年底,至少有11款源自中国本土研发的创新药获得了FDA批准并在美国上市。[14] 下面简要回顾其中具有代表性的案例:
泽布替尼(Zanubrutinib):由百济神州开发,用于治疗套细胞淋巴瘤(MCL)。这是首个中国自主研发的小分子靶向药在美国加速批准上市的案例(FDA批准时间2019-11-14)。值得一提的是,泽布替尼的关键注册研究以中国主导的单臂临床试验 为基础,由于MCL属于罕见疾病且已有替代疗法有限,FDA在缺乏充分美国患者数据的情况下获准上市。泽布替尼在中国上市晚于美国(2020-06-02),基于相同的关键临床试验。
艾贝格司亭α(Efbemalenograstim alfa):由亿一生物开发,用于肿瘤患者的中性粒细胞减少症。该产品在2016年启动了一项国际多中心III期研究(含美国,无中国受试者)。在2018年4月,同时在中国和国际多中心(含美国,无中国)分别开展了一项III期研究。2023年中国和美国分别基于2项III期临床试验结果,同年批准了艾贝格司亭α上市。
舒沃替尼(Sunvozertinib):由迪哲生物开发,用于既往经治的EGFR 外显子插入突变的局部晚期或转移性非小细胞肺癌。这是最新FDA批准的中国创新药(2025-07-02)。在I临床时,开展的国际多中心研究。在美国获批的关键临床(国际多中心,WU-KONG1)开展的时间(2019-07-09),比中国批准的关键临床(中国多中心,WU-KONG6)要早(2021-07-19)。推测受试验设计 ,以及入组速度的影响,中国的先完成(WU-KONG1设置了2个剂量组,目标入组数量更大)。
表格 5 中国创新药在美国上市药品列表
药品
美国
上市日期
中国
上市日期
MAH(中国)
作用机制
适应症
关键临床
研究编号
设计
受试者人数
泽布替尼
2019-11-14
2020-06-02
百济神州
BTK抑制剂
套细胞淋巴瘤
中国多中心
BGB-3111-206
2期,多中心,单臂,开放标签
86
瑞马唑仑
2020-07-02
2019-12-26
江苏恒瑞
苯二氮䓬类药物
镇静
美国多中心
NCT02290873
NCT02296892
NCT02532647
3项3期临床,多中心,随机 双盲,平行对照/安慰剂 对照/阳性对照
966
本维莫德
2022-05-23
2019-05-29
广东中昊药业
芳香烃受体激动剂
银屑病
美国+加拿大多中心
DMVT-505-3001
DMVT-505-3002
2项3期临床,随机,双盲 ,安慰剂对照
1025
特瑞普利单抗
2023-10-27
2018-12-17
上海君实生物
PD-1单抗
鼻咽癌
区域多中心(中国大陆+新加坡+台湾地区)
NCT03581786
1项3期临床,随机,安慰剂对照
289
呋喹替尼
2023-11-08
2018-09-04
和记黄埔
VEGFR激酶抑制剂
结直肠癌
国际多中心(包含美国)
FRESCO-2
1项3期临床,随机,双盲,安慰剂对照
691
艾贝格司亭α
2023-11-16
2023-05-06
亿一生物
人G-CSF和人IgG2 Fc片段组成的重组融合蛋白
中性粒细胞减少症
国际多中心(包含美国)
NCT02872103
NCT03252431
2项3期临床,随机,双盲,安慰剂对照;
随机,开放标签,阳性对照
515
替雷利珠单抗
2024-03-13
2019-12-27
百济神州
PD-1单抗
食管鳞癌
国际多中心(包含美国)
NCT03430843
1项3期临床,随机,对照,开放标签
512
恩沙替尼
2024-12-18
2020-11-17
贝达药业
ALK 抑制剂
非小细胞肺癌
国际多中心(包含美国)
NCT02767804
1项3期临床,随机,开放标签,阳性对照
290
派安普利单抗
2025-04-23
2021-08-03
正大天晴康方
PD-1单抗
鼻咽癌
国际多中心(包含美国)
中国多中心
NCT04974398
NCT03866967
1项3期,随机,双盲;
1项2期,开放标签,单臂
416
他雷替尼
2025-06-11
2024-12-25
葆元生物
酪氨酸激酶抑制剂
非小细胞肺癌
中国单中心
国际多中心(包含美国)
NCT04395677
NCT04919811
2项2期,单臂,开放标签
270
舒沃替尼
2025-07-02
2023-08-22
迪哲医药
EGFR酪氨酸激酶抑制剂
非小细胞肺癌
国际多中心(包含美国)
NCT03974022
1项2期,单臂
85
表格 5所列FDA批准的中国创新药显示出一个显著趋势:除早期以中国主导数据获批的个案(如泽布替尼)外,近年来绝大多数产品的关键注册试验均为国际多中心设计,且明确涵盖美国受试者 。FDA在临床评估中对本土数据的重视程度不断提升,也促使中国企业在早期研发阶段就更主动地规划全球数据生成策略,特别是在关键适应症和主流治疗领域。
泽布替尼作为首例基于中国数据在美获批的小分子靶向药,其特殊性与罕见病定位、替代治疗有限及Project Orbis通道密切相关。然而后续产品,包括呋喹替尼、艾贝格司亭α、恩沙替尼等,均通过涵盖美国的国际多中心研究作为核心数据支持,在临床设计上更贴近FDA对多地区、多人群可推广性的基本要求。这一转变说明,中国企业在经验积累与监管理解深化后,已逐步形成面向美国市场的标准化开发路径。
总体来看,表中所列产品代表了中国创新药从区域经验走向国际共识的关键节点。未来中国企业若希望进一步拓展海外市场,特别是进入欧美主流监管体系,早期纳入目标国人群、采用国际规范的临床设计、以及主动对接多国监管将成为基础要求而非例外路径。
五、讨论
综上所述,中美两国在创新药物监管方面正日趋接轨。从进口创新药角度看,中国通过改革药品注册分类和接受境外临床数据,大大加快了新药引进步伐,2015–2025十年间中美新药上市时差明显缩短,“同步上市”正逐渐成为现实。大量国际多中心试验将中国纳入,使得中国患者在全球新药开发中占有一席之地,这不仅利于中国加速审批,也提高了试验数据的普适性。然而在特定领域和特定药物上,桥接试验仍扮演重要角色,尤其当涉及种族差异或本土医疗需求时,中国监管机构通过灵活运用小规模桥接和附条件批准,平衡了新药引进的速度与安全性考量。
从国产创新药出海的视角观察,过去的十年间中国创新药企逐步突破国际门槛,但目前仍集中在肿瘤及恶性血液病领域。成功经验表明,高质量的临床数据和正确的研发策略是赢得FDA认可的关键。中国药监部门以鼓励创新和加速审批的姿态,欢迎本土新药“走出去”并同步“走回来”。可以预见,随着中国全面加入ICH 并不断更新本国监管指南,中美两国的新药研发和审批将更加同步化、标准化。未来我们将看到更多创新药物实现中美同步上市,中国创新药的国际地位也将持续提升。良性循环终将使全球患者受益,加速创新药物惠及更广泛的人群。
参考文献:
1. 整理自 Novel Drug Approvals at FDA:url
2. 相应新分子实体 首次在中国获批,不限定为原研进口。
3. 4个产品按照化药3类(注册分类改革后:境内申请人 仿制境外上市但境内未上市原研药品的药品。该类药品应与参比制剂 的质量和疗效一致)在中国批准上市,分别为利非司特(成都康弘)、布立西坦(江西科睿)、司替戊醇(石家庄四药)、奥赛利定(江苏恩华)。
4. 兰地洛尔(南京海辰)为旧注册分类 化药3.1(已在国外上市销售的制剂及其原料药 ,和/或改变该制剂的剂型,但不改变给药途径的制剂)在中国获批上市,申报规格、适应症与原研产品(商品名Onoact)一致,参见《注射用盐酸兰地洛尔(CXHS1500147)申请上市技术审评报告》 ;信达生物的替妥尤单抗N01,与原研替妥尤单抗(商品名Tepezza)氨基酸序列、作用靶点、给药途径 、给药剂量 和适应症相同,在剂型 上进行了改良,原研为冻干粉针,替妥尤单抗N01为注射液 ,参见《替妥尤单抗N01注射液(CXSS2400050)申请上市技术审评报告》 。
5. CDE公布审评卷宗的有2个产品:仑伐替尼在中国递交上市申请时,拟定适应症为肝细胞癌,当时FDA尚未批准相应适应症;乌帕替尼在中国递交上市申请时,拟定适应症为特应性皮炎,当时FDA尚未批准相应适应症。
6. 参考注释3。
7. 参考注释4。
8. 参考注释4。
9. 与化药2.4类似,CDE公布审评卷宗的有3个产品:德曲妥珠单抗在中国递交上市申请时,拟定适应症为至少接受过1种或1种以上抗HER2治疗的不可切除或转移性HER2阳性乳腺癌,当时FDA批准的适应症为至少接受过2种或2种以上抗HER2治疗的不可切除或转移性HER2阳性乳腺癌;维泊妥珠单抗在中国递交上市申请时,拟定适应症为联合利妥昔单抗、环磷酰胺、多柔比星和泼尼松适用于治疗既往未经治疗的弥漫大 B 细胞淋巴瘤,当时FDA批准的适应症为联合苯达莫司汀和利妥昔单抗用于治疗既往至少接受过两种治疗后复发或难治性的弥漫大 B 细胞淋巴瘤;比奇珠单抗 在中国递交上市申请时,拟定适应症为活动性强直性脊柱炎,当时FDA批准的适应症 为斑块型银屑病。
10. 截止到2026.1.14。
11. 这里的分析基于有CDE公开审评资料的进口创新药(不包含国产国产创新、国内仿制或改良产品),总数是125。国际临床试验:根据审评资料里有效性分析的临床试验,临床分期统计按照关键临床的最高分期进行统计;审评卷宗中未明确国际临床中是否包含中国人,则按照无中国人入组统计。国内桥接:根据审评资料里所有在国内开展的桥接临床试验(包括PK、安全性、有效性),临床分期按照所有临床试验中分期最高的统计。
12. 本分析基于中国国家药监局药品审评中心(CDE)公开的审评资料,对125个进口创新药的临床试验 数据进行了整理。需要强调的是,这一样本并不涵盖所有进口创新药,因而所观察到的趋势具有一定局限,结论应谨慎解读。此外,数据来源于监管审评公开信息,旨在客观反映不同药物在获批前是否有中国患者参与国际多中心临床试验 ,以及是否在中国开展了附加的桥接研究 。
13. 无临床相关要求”指的是,不需要在临床数据方面有要求,可能只需要完成其他领域或是药物警戒 要求。“完善临床数据”指的是上市申请时提交的临床数据 以作为证据支持上市申请,当仍需继续跟踪数据,或者一些进口创新药在美国上市的时候,FDA已经要求开展确证性临床 等,企业已经计划和开展,需要继续完善数据。“开展相关研究”指的是中国监管机构对企业额外要求开展的在中国本土的研究。
14. CDER批准,未涵盖CBER批准的(传奇的CART)。
识林-木兰
识林® 版权所有,未经许可不得转载
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保临床试验数据的合规性与完整性,与药审中心沟通以确保临床试验设计符合中国药品注册要求。 注册:在药品注册申请中,使用境外临床试验数据时,需确保数据的完整性和符合性,并与药审中心进行有效沟通。 临床:在设计和执行临床试验时,需遵循ICH GCP标准,确保数据的科学性和准确性。 文件适用范围:
文件要点总结:
数据真实性与完整性: 强调境外临床试验数据必须真实、完整、准确和可溯源,符合ICH GCP要求。设计与质量管理体系: 申请人需确保临床试验设计科学,质量管理体系符合要求,数据统计分析准确完整。数据提交要求: 境外临床试验数据用于中国注册申请时,需提供所有相关数据,不得选择性提交。技术文件格式: 鼓励采用通用技术文件格式(CTD)提交临床试验数据,包括生物药剂学、临床药理学、有效性和安全性资料。数据可接受性评估: 根据数据质量,境外临床试验数据可被完全接受、部分接受或不接受,部分接受时需与药审中心沟通并可能需开展额外临床试验。以上仅为部分要点,请阅读原文,深入理解监管要求。