适用岗位:
- QA(质量保证):必读。应根据文件更新质量控制流程,特别是关于除菌过滤的参数和风险管理。
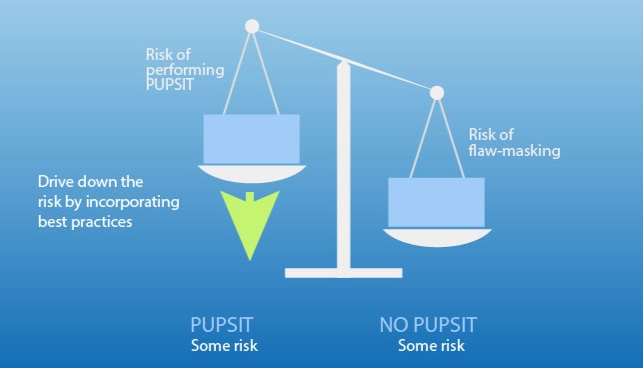
- 生产:必读。需确保生产过程中遵循除菌过滤的最新要求,特别是在实施PUPSIT时的操作。
- 研发:必读。在设计过滤系统和验证过程中,需考虑文件中的指导原则。
- 注册:必读。在药品注册过程中,需确保申报资料符合最新的GMP要求,特别是关于除菌过滤的部分。
工作建议:
- QA:更新SOPs以符合PUPSIT和冗余过滤的最新要求,并对相关人员进行培训。
- 生产:在实施PUPSIT时,注意压力、流速和时间的控制,确保操作符合Annex 1的规定。
- 研发:在设计过滤系统时,考虑PUPSIT的实施对验证过程的影响,并选择合适的过滤参数。
- 注册:确保注册文件中包含有关除菌过滤的详细描述,符合EU GMP的最新要求。
适用范围:
本文适用于欧盟GMP指南附录1中关于化学药品的无菌过滤实践,特别针对创新药和仿制药的生产,由欧盟发布,适用于Biotech、大型药企和跨国药企。
要点总结:
欧盟GMP指南附录1强调了在整个生产过程中质量风险管理的重要性,特别是在无菌过滤操作中。文件详细描述了无菌过滤的操作要求,包括预使用后灭菌完整性测试(PUPSIT)的重要性,以及在实施PUPSIT时可能面临的挑战。讨论了PUPSIT对验证过滤过程的影响、使用冗余过滤降低过滤掩蔽风险的可能性,以及在不同情况下选择水或产品作为PUPSIT的湿润溶液。此外,还探讨了在细菌保留测试中评估PUPSIT最大压力的必要性,以及在生物负荷采样和替代品使用中的最佳实践。这些指导原则旨在确保最终产品的无菌性,并为制造商提供实施挑战的具体指导。
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
- QP(Qualified Person):应熟悉远程批认证的条件与技术要求,确保在执行远程批认证时符合GMP要求。
- 生产部门:必须理解无菌药品生产、ATMP生产的特殊GMP要求,以及共享生产设施的交叉污染风险控制。
- 质量保证部门(QA):需掌握GMP和GDP的问答内容,确保药品生产和分销的质量控制。
- 注册部门:应了解活性物质注册要求,以及GMP证书、不符合声明和生产许可的相关信息。
文件适用范围:
本文适用于人类和兽医药品,涵盖了化学药、生物制品、疫苗和中药等药品类型。针对创新药、仿制药、生物类似药、原料药等注册分类,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。由欧洲药品管理局(EMA)发布,遵循欧盟(EU)的GMP和GDP指南。
文件要点总结:
- 远程批认证/确认:更新了关于QP远程批认证的条件,包括技术要求和签名的最小要求。
- QP居住地要求:2023年7月新增内容,讨论了QP是否需要居住在授权站点所在的成员国。
- 无菌药品生产:强调了无菌药品生产的补充要求,包括对生产环境和过程的严格控制。
- ATMP生产:提供了针对先进疗法药品(ATMP)的GMP特定要求,包括生物来源起始物料的生产原则。
- 活性物质注册:详述了用于人类药品的人用活性物质的注册要求,包括制造商和进口商的义务。
以上仅为部分要点,请阅读原文,深入理解监管要求。