制剂(DP)的成分列表与处方组成方面,FDA的灵活性在于豁免确立最终的或商业化的剂型与处方工艺。即使在申报时申办者采用的是诸如粉末装入胶囊(Powder in capsule)或粉末装入药瓶(Powder in bottle)这类仅含DS、不含任何辅料的极简制剂形式,FDA通常也能接受,但在技术审评时更依赖DS数据。
与DS类似,对于DP的质量控制与分析方法,FDA通常不需要申办者针对原料药工艺特异性杂质(DS process specific impurities)以及那些在制剂生产过程中不会受到影响的质量属性进行重复控制。申办者在此阶段也不需要提交针对制剂的元素杂质和亚硝胺杂质风险评估报告。此外,分析方法同样不需要提供验证数据,并且如果制剂中没有观察到新的降解产物,IND中也只需进行简单陈述即可。FDA充分理解并接受制剂的质量标准在研发早期会随着工艺优化而动态变更,申办者可以使用代表性批次的质量标准和批数据来证明其具备生产合格产品的能力。
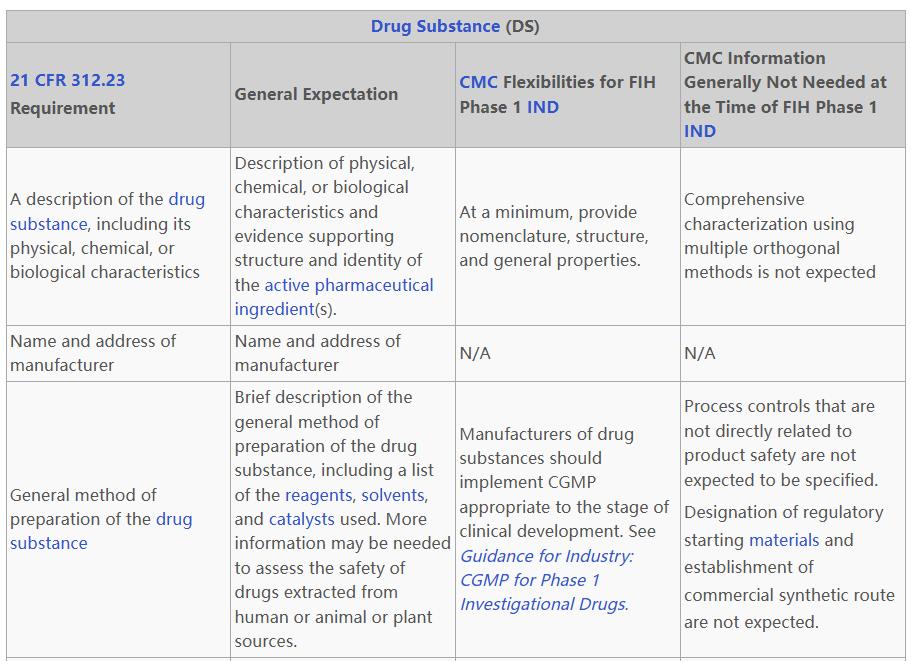
原液(DS)制备工艺方面,除了明确申办者可采用相称的CGMP,且不需要指定与产品安全非直接相关的工艺控制指标,FDA的灵活性还体现在细胞库与病毒安全性评价上:申办者可无需确证细胞库的克隆源性(Derivation of clonally derived cell bank is not expected),也不需要建立传统的两级细胞库(Two-tiered cell bank)系统;同时,对于细胞库或未处理收获液(UPB)的体外外源因子检测,不要求进行21天或28天培养的扩增步骤。此外,如果申办者能够合理利用现有的先验知识(Prior Knowledge),FDA不要求提供产品特异性的病毒清除研究结果。
DS的质量控制与分析方法方面,FDA放宽效价测定与方法验证。对于已被证明不具有高级结构(Higher order structure)的简单蛋白质,申办者不需要进行效价测定;若质量控制方法的开发在IND申报前无法完全就绪,申办者也可以仅使用效价表征结果(Potency characterization results)来启动FIH Phase 1临床试验。在方法与标准的成熟度上,FDA在此阶段不要求提供分析方法验证数据,且通常不需要建立反映作用机制(MOA)的细胞水平效价测定。此外,申办者也不被要求建立两级参考标准品(Two-tiered reference standard)系统。