首页
>
资讯
>
FDA 修订临床主方案指南草案,澄清更多技术细节
出自识林
FDA 修订临床主方案指南草案,澄清更多技术细节
2026-07-08
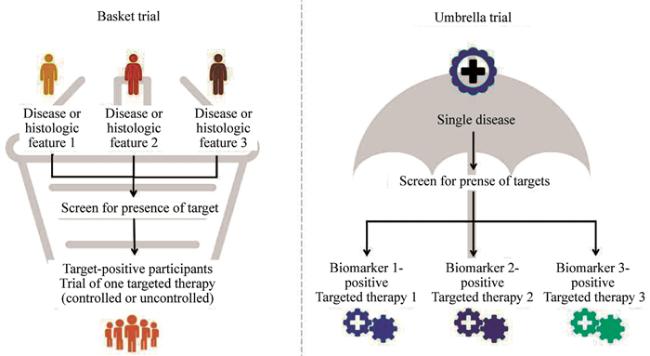
6月22日,FDA发布修订版指南草案《药品和生物制品研发主方案》 ,替代2023年12月发布的同名草案 。除已有的伞式试验(Umbrella Trial),修订版草案主要新增篮式试验 (Basket trial)的相关建议,并在随机化 、对照选择、知情同意 、盲法 设计、信息借用以及IND 申报框架等方面作出了澄清性修订。
本指南修订也是FDA响应美国卫生部(HHS)于6月22日发起的“临床试验开拓者行动” (Operation TrialBlazer)计划的系列行动之一。
以下为部分要点概览,仅供参考,技术细节以原文为准。
篮式试验信息借用
指南III.G小节中将篮式试验跨子研究的信息借用(borrowing)描述为一条统计学连续谱(continuum),一端为完全不借用数据的独立分析,另一端为全人群合并分析,中间则为基于贝叶斯分层模型(bayesian hierarchical models)等依据疗效相似度动态借用的定量方法。信息借用程度越高,对某一子人群疗效估计的偏倚 可能随之增大,因此申办者必须以操作特性(operating characteristics)评估来论证所选统计方法在不同疗效异质性场景下的假阳性、假阴性概率及偏倚-方差权衡。
申办者还需评估病理生理相似度、药物作用机制及药代特性、终点指标及其他估计目标 的属性相似性。在科学论证充分的前提下,主方案中目标疾病子研究可作为“充分且对照良好的临床试验”,其他相关子研究的结果可作为确证性证据,共同支撑“实质性证据 ”的认定。关于“实质性证据”,可参考同日发布的《人用药品和生物制品有效性的实质性证据的证明》 修订版指南草案 。
随机化与对照组重构
“随机化”小节明确方案选择应综合考虑效率与操作因素,区分“各药与对照比例恒定”(constant randomization ratio between each drug and control)和保持“被分到对照组 的概率恒定”(constant probability of being randomized to control over time)的随机化思路。允许符合条件的参与者在清洗期 (washout period)后重新入组。强调必须在统计分析中妥善处理同一参与者产生的相关性数据。
“对照组”中引入“同期合格对照参与者(concurrently eligible control participants)”概念,即满足入组标准、且本可被随机至该药、但被同期随机至对照的参与者。对于旨在提供有效性实质性证据的主方案,申办者一般应仅采用同期合格对照。使用非同期对照或不符合合格标准的同期对照属“罕见情形”,须在试验启动前与 FDA 协商并达成一致。
盲法与知情同意
“盲法”补充在给药途径 /方案相似的药物组内使用多重模拟(multiple-dummy)联合共享对照的设计,以及对部分盲法(partial blinding)下安全性结局分析的交叉引用(详见 III.I);存在偏倚担忧时建议改用完全盲法,或仅以匹配对照作主分析。
“知情同意”中建议申办者缩短知情同意书 篇幅并提高可读性。明确指出“两步知情同意(two-step consent,即在随机化后再签署特定子研究知情同意书)”会引入潜在偏倚。此时申办者需提供充分论证并描述拟采取的缓解偏倚措施。
IND申报框架
FDA 建议申办者在提交主方案IND 前申请Type B Pre-IND 会议,因主方案复杂度较高,常需多轮会议沟通。
对于部分篮式试验和带有篮式元素的平台试验,当各子研究所需的审评专业知识分布在多个审评部门时,可单独提交子研究IND。
指南草案分别规定了“全部子研究在主方案IND下提交”和“单独提交子研究IND”两种路径下的方案更新流程,要求各路径均保持主方案IND与所有子研究IND之间的交叉引用和变更 通知。
作者:识林-白蜡
识林® 版权所有,未经许可不得转载
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位及建议: 注册 :梳理现有试验数据与指南要求的匹配性,提前规划确证性证据来源。 临床 :优化试验设计(如终点选择、对照组设置),确保符合“充分且良好对照”标准。 研发 :在早期阶段(如Ⅱ期结束前)与FDA讨论机制研究或自然病史数据作为确证性证据的可行性。 医学 :评估疾病严重性和未满足需求,为监管灵活性申请提供科学依据。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
临床团队(必读) :优化随机化和对照组设计,确保盲法实施;提前与FDA讨论非同期对照组使用的合理性。 统计团队(必读) :评估多重性影响,预设跨亚组分析的统计方法;验证非同期数据整合的偏倚控制策略。 注册团队(必读) :主导IND提交,协调跨IND引用;建立主方案与子研究的电子提交标签系统(eCTD)。 安全团队 :制定统一的不良事件收集标准,区分部分盲法下的药物特异性安全分析。 以上仅为部分要点,请阅读原文,深入理解监管要求。