首页
>
资讯
>
FDA 药品质量状况年报:生产持续外流,海外检查比例下降
出自识林
FDA 药品质量状况年报:生产持续外流,海外检查比例下降
2026-06-29
6月25日,FDA药品审评与研究中心(CDER)发布《2025财年药品质量状况报告》 ,这是该系列报告的第8版(历史版本可至识林查阅)。报告基于CDER场地名录、检查数据、缺陷 信息等多维度信息,系统呈现了来自全球场地的药品生产质量的最新态势。
报告显示,全球药品生产网络仍在经历从美欧向中印的结构性转移,美国本土生产场地数量持续缩减,而中国和印度的场地份额稳步上升。中国场地的官方行动指示(OAI)比例低于全球平均水平,美国的OAI比例则明显偏高。此外我国出海药企还需注意的是,场地历史遗留问题可能导致药品申请遭拒批。
生产场地分布:美国净减5%,中印份额持续上升
截至2025财年末,CDER场地名录收录全球5953个药品生产场地,其中57%位于美国。需要注意的是其中包括占美国场地50.3%(1698个)的医用气体 生产场地。
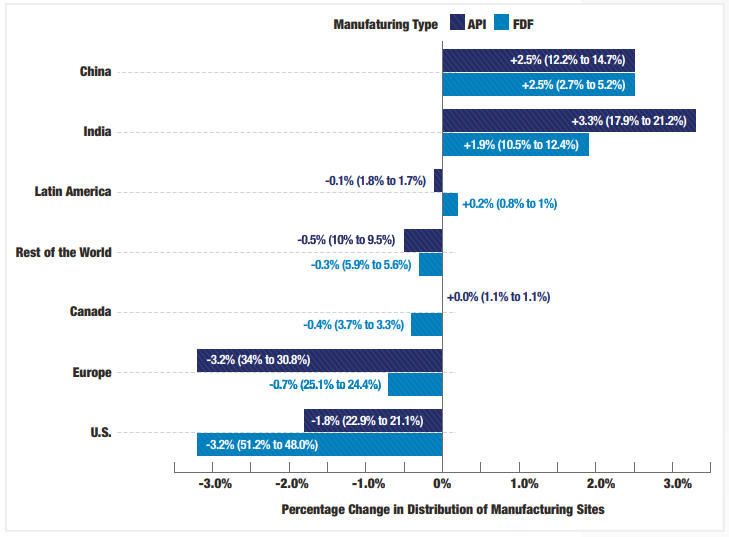
CDER场地名录中的场地数量从2021财年的5999个降至2025财年的5953个,降幅1%。同期,美国场地数量净减少5%。过去五年中,印度、中国、西班牙、德国、意大利和瑞士的场地净增长率最高,合计增长超过10%。
总体趋势显示,生产分布从美国和欧洲向中国和印度转移。对于原料药(API) 生产场地,中国的分布份额增加2.5%,印度增加3.3%,而欧洲和美国分别下降3.2%和1.8%。成品制剂(FDF)生产场地的分布变化呈现类似趋势。报告指出,这些变化主要归因于中国和印度生产场地的增加,而美国和欧洲的场地数量相对稳定。
美国白宫和FDA在2025年启动了多项生产“回流”举措 ,其成效尚待时间检验。
检查数据:重心在美国境内,中国检查结果相对较好
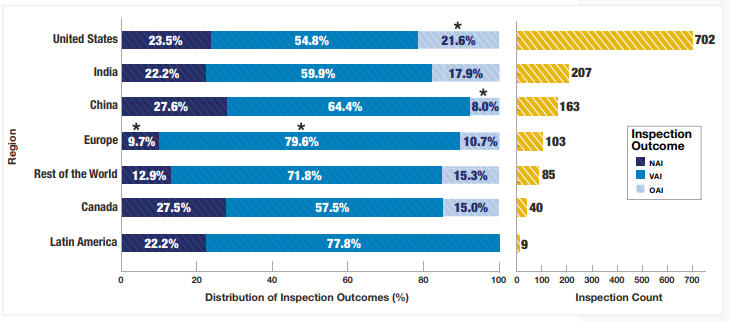
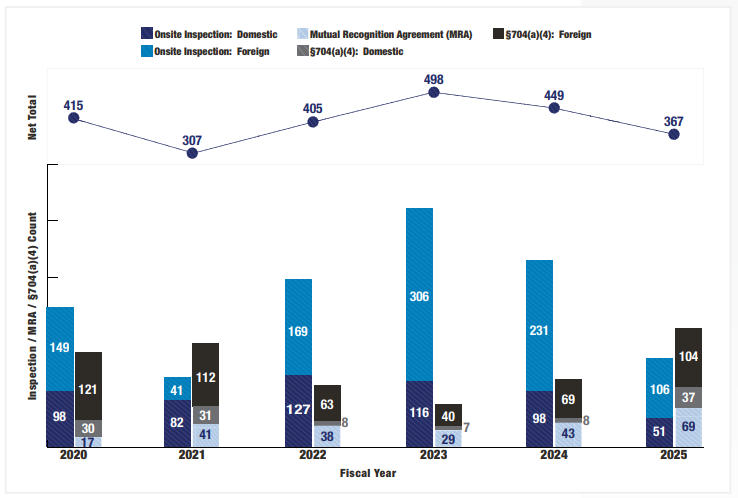
2025财年,CDER进行了1,248次药品质量保证 检查,其中702次(56%)在美国,546次(44%)在海外。加上61次互认协议(MRA)伙伴检查,总计1,309次检查。相比之下,2024财年进行了972次检查,其中432次(44%)在美国,540次(56%)在海外。可见检查总数上升,海外检查的数量也略增,但比例却在下降,显示检查更多侧重美国国内。
检查结果显示,22%为无行动指示(NAI),60%为自愿行动指示(VAI),18%为OAI。这其中中国的OAI结果最少(8.0% vs 18%全球平均水平),美国的OAI结果较多(21.6% vs 18%全球平均水平),欧洲的NAI结果少,VAI结果显著多于全球平均水平。此外,中国的NAI比例也是全球最高,加拿大紧随其后。
上述数据似乎可以读出FDA的海外检查次数仍受限于资源,尽管受到国会和政府的持续压力,已启动多项举措 ,但未能看到明显的趋势变化。
PAI和PLI:更频繁使用MRA和记录索取
作为新药申请(NDA) 、生物制品许可申请(BLA) 和简化新药申请(ANDA) 审批流程的一部分,FDA考察产品风险和生产风险,以及申报材料的准确性和可靠性,从而决定是通过批准前检查(PAI) 或许可前检查(PLI)、记录索取(Record Request),还是利用FDA现有信息或“互认协议”(MRA)合作机构检查信息来评估。按FDA的“替代工具”指南 ,远程监管评估(RRA)也是预先评估工具之一,但报告中未涉及。
2025财年,FDA使用了创纪录的69次MRA合作机构检查来支持PAI/PLI场地评估。FDA与欧盟、英国和瑞士的MRA分别自2017年、2021年和2023年生效。
2025财年有141次记录索取审查用于支持基于风险的申请决策,这是自2021财年以来最多的一年。其中74%发送给海外生产商,发送给印度和中国的比例最高(分别为21%和12%)。
虽然BLA仅占2025财年FDA批准申请的4.5%,但它们占记录索取审查的42%(141次中的59次)。
场地历史遗留问题是申请拒批原因之一
FDA对2024-2025财年期间收到的近10000份需要场地评估的原始申请和补充申请 进行了分析。截至2026年5月,28%的申请收到了完全回应函(CRL) 。其中,43%的CRL(占所有申请的12%)源于“场地暂停”(facility withhold)。可供参考的是,识林此前报道相当数量新药CRL源自质量问题 。
进一步分析发现,超过50%的场地暂停与申请提交时已存在的OAI或潜在OAI(pOAI)状态直接相关——即这些场地在申请前就存在历史遗留的合规问题。其余场地暂停则主要源于审评周期内现场检查所发现的缺陷,导致OAI/pOAI。
可见,当前的OAI/pOAI状态在这些申请中的高频出现,显著增加了因场地问题而收到CRL的风险。FDA指出,申办者应将生产场地的合规状态作为成功申报的关键因素之一。
导致场地暂停的前三大检查缺陷包括:
调查不充分:未能彻底审查生产记录中的偏差 或批次失败;
质量管理部门 (QCU)职责缺失:相关职责或程序未书面规定,或未按规定执行;
作者:识林-实木
识林® 版权所有,未经许可不得转载。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。