首页
>
资讯
>
ICH 里约大会批准使命愿景和战略规划,定稿 GCP,多指南取得进展
出自识林
ICH 里约大会批准使命愿景和战略规划,定稿 GCP,多指南取得进展
2026-06-09
ICH 于6月2-3日在巴西里约热内卢召开半年度大会。大会批准巴基斯坦DRAP和新西兰Medsafe成为观察员。至此,ICH共有25个成员和43个观察员。
大会批准了《ICH 战略愿景》 ,概述了通过协调指南推进公共健康的愿景、使命和作用。大会还审议并批准了《ICH 协会2025年度报告》 和《ICH技术工作组最终报告》 。下次ICH半年度会议将分别于2026年11月14-18日在捷克布拉格、2027年6月12-16日在西班牙马德里举行。
ICH新指南进展:定稿GCP,多份指南按计划推进
大会通过了ICH《E6(R3) 药物临床试验质量管理规范 - 附件2》 最终版本。值得一提的是,NMPA也刚于昨天(6月8日)定稿中国版GCP ,此前征求意见稿起草说明 就已明确两份GCP 之间的关系:“E6(R3)中属于建议性做法和理念性描述,而非监管刚性要求的不纳入我国GCP,而是采取全文实施E6(R3)方式,为临床试验 实施灵活性预留空间”。
对于在开发中的指南,12个ICH专家组同期在里约热内卢举行面对面会议。大会收到各工作组报告,确认下列广泛领域的ICH指南取得进展(可至“ICH索引 ”查看对应文件):
药典讨论组报告了ICH Q4B 维护活动进展。
此外,监管活动医学词典(MedDRA )29.0版已于2026年3月以27种语言发布,伴随文档4.0版预计今年晚些时候发布。维护和支持服务组织(MSSO)于2025年10月推出新下载API。培训已覆盖108个国家超过16,000名用户,拉美、亚洲、非洲和中东地区参与度加强。
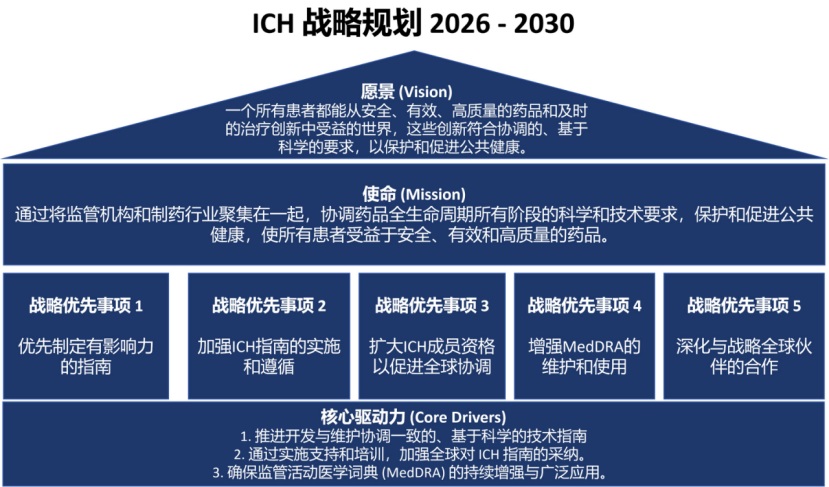
以下是《ICH 战略愿景》 中部分原文翻译:
ICH的VVM(价值、愿景和使命)
我们的价值主张
ICH是一个全球非营利组织,其独特之处在于将监管机构和制药行业聚集在一起,协调药品安全、有效性和质量的科学和技术要求,造福各地患者。
ICH召集各领域世界顶尖专家制定协调指南,这些指南被视为药品安全、有效性和质量的全球标准,并嵌入世界许多司法管辖区的监管框架中。
ICH协调指南促进行业更一致和精简的提交,以及监管机构更及时和连贯的评估,有助于更快地为患者提供安全、有效和高质量的药品。相应地,全球协调和监管一致性支持治疗创新。
我们的愿景
一个所有患者都能从安全、有效和高质量的药品以及及时的治疗创新中受益的世界,这些创新符合协调的、基于科学的要求,以保护和促进公共健康。
我们的使命
通过将监管机构和制药行业聚集在一起,协调药品全生命周期 所有阶段的科学和技术要求,保护和促进公共健康,使所有患者受益于安全、有效和高质量的药品。
ICH的战略优先事项
此处提出的五个战略优先事项为ICH在其规划和开展核心活动中提供框架,并履行其通过全球协调的药品技术和科学要求保护和促进公共健康的使命。
优先制定有影响力的指南
ICH将努力在最能推进公共健康、药品可及性、科学创新和药品高效全球开发的领域优先进行协调。ICH指南应具有影响力,支持创新,并跟上科学进步、技术发展和工艺革新。此外,ICH将提高指南制定流程的效率,包括更充分利用数字平台和工具,并有选择性地应用人工智能 。
加强ICH指南的实施和遵循
充分实施ICH指南是实现监管要求全球协调的前提,支持更快、更可预测的全球药品可及性,保护患者安全,提高药品质量和数据可靠性 ,支持高效的药品开发和评估,确保监管机构的透明和基于科学的期望,最终支持药品安全标准和公众信任。ICH将通过培训、实施监测和应对实施挑战,支持监管成员对ICH指南进行连贯、一致和充分的实施。
扩大ICH成员资格以促进全球协调
ICH将努力扩大成员资格和拓展外联工作,以最大化ICH指南的全球采用。扩大成员资格对于增加安全、有效和高质量药品要求科学一致的全球覆盖范围至关重要。对于监管机构而言,加入ICH带来价值并展示监管成熟度和诚信度,这也增加了开发和维护协调指南所需专业知识的获取途径。
增强MedDRA的维护和使用
ICH将确保MedDRA跟上科学和医学进步的步伐,满足世界各地用户的不断变化的需求,包括通过及时更新、高完整性、多语言访问以及与其他术语的互操作性。在药品许可之前和之后,MedDRA都是药品注册、文档和安全监测的强大工具。ICH将寻求确保MedDRA作为监管信息交流使用的主要术语,并支持MedDRA在全球范围内的采用,以更好地保护患者健康。
深化与战略全球伙伴的合作
ICH寻求机会,以更好地加强与相关国际组织的接触与合作,从而扩大对ICH指南的理解、采纳及其影响力。ICH将制定一个战略框架,以指导互利合作的发展,包括以谅解备忘录为约束的合作。全球合作伙伴可包括,例如,代表患者的组织,或专注于国际标准制定和培训交付的组织。
作者:识林-实木
识林® 版权所有,未经许可不得转载。
适用岗位及工作建议 必读岗位 :
临床开发(Clinical) :需将患者偏好纳入临床试验设计,优化终点选择和招募策略。 注册(RA) :确保PPS数据符合监管提交要求,协调CTD模块整合。 市场准入(Market Access) :利用偏好数据支持产品差异化策略和定价论证。 患者参与(Patient Engagement) :主导PPS前期调研和患者咨询,确保研究设计反映真实需求。 适用范围 本文适用于全球(ICH成员地区)化学药、生物制品及疫苗的研发与评价,涵盖创新药、生物类似药及预防/诊断类产品。适用企业包括跨国药企、Biotech及CRO/CDMO,重点关注治疗领域,但延伸至健康人群的预防性干预。
文件概要 该指南阐述了患者偏好研究(PPS)在药物开发全周期中的应用框架,涵盖定性(访谈、焦点小组)与定量(离散选择实验等)方法。指南指出PPS可识别未满足需求、优化临床试验终点权重、评估获益-风险权衡阈值,并支持监管决策(如ICH M4E(R2)中的获益-风险评估)。研究设计需遵循多学科协作原则,强调预测试、样本代表性和数据质量控制,如通过认知访谈验证工具有效性。
PPS结果需整合至CTD模块2(如2.5.1开发依据)和模块5(5.3.5.4临床研究报告),并可能用于定量获益-风险分析。指南建议早期与监管机构沟通以确保方法合规性,同时允许跨区域数据引用,但需论证本地适用性。
以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: 注册 :评估RWD适用性,确保数据源符合监管要求;修订协议以纳入分散化元素的操作细节。 临床运营 :设计远程访视流程,制定试验产品配送的应急方案;培训研究者使用数字健康技术(DHT)。 数据管理 :建立多源数据整合规则,验证RWD的完整性与一致性;实施数据去标识化措施。 QA :更新SOP以覆盖分散化试验的监控要求,定期审计服务商的数据隐私合规性。 安全监测 :优化远程安全性数据收集机制,确保研究者及时获取关键信息。 以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
生物等效性研究人员:深入理解并应用本指南进行生物等效性研究设计和数据分析,确保研究的科学性和合规性。 临床研究部门:在设计临床试验时,考虑本指南对受试者选择、研究设计、样本量计算等方面的要求。 质量管理(QA):确保生物等效性研究的执行和记录符合本指南的规定。 注册部门:在药品注册过程中,依据本指南准备和提交生物等效性研究资料。 文件适用范围:
要点总结:
研究目的与背景 :明确了生物等效性研究的目的,即通过药代动力学终点评估口服固体制剂的生物等效性,并强调了数据完整性的重要性。研究设计原则 :推荐使用随机、单剂量、交叉研究设计,并详细说明了研究人群选择、样本量计算、研究设计和条件等关键要素。数据与统计分析 :强调了非重复研究设计的数据呈现和统计分析方法,包括生物等效性分析人群的考虑、数据呈现、统计分析的一般考虑等。特殊主题 :讨论了内源性化合物、其他速释剂型(如口腔崩解片、咀嚼片、口服悬浮液)、固定剂量组合和pH依赖性等特殊主题。文档记录 :要求生物等效性研究报告应包含完整的研究协议、执行和评估记录,并符合ICH E3的格式。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必读。需根据M4Q(R2)指南更新质量管理体系,确保所有适用的ICH和区域指南被正确实施,并监督合规性。 注册(Regulatory Affairs):必读。负责根据M4Q(R2)指南准备和提交注册文件,确保申报资料符合ICH和目标注册国家的要求。 研发(R&D):必读。需依据M4Q(R2)指南进行药品开发,确保开发过程中的关键质量属性(CQAs)被合理控制,并在申报资料中体现。 生产(Production):必读。应根据M4Q(R2)指南优化生产流程,确保生产过程和控制策略能够持续生产出符合质量要求的药品。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
注册(RA) :必读。需了解ICH M7 R2 Addendum的最新要求,以确保公司药品注册文件符合最新的国际标准。研发(R&D) :必读。在新药研发过程中,需参考该文件中关于化合物特异性可接受摄入量的计算方法,以评估和控制药物中的遗传毒性杂质。质量保证(QA) :必读。需掌握文件中的合规要求,确保质量控制流程和产品规格符合ICH指南。临床(Clinical) :必读。在临床试验设计和执行过程中,需参考文件中关于化合物风险评估的原则,以保护受试者安全。工作建议:
注册(RA) :应及时更新注册文件模板,确保所有注册活动符合ICH M7 R2 Addendum的要求,并与监管机构沟通确认。研发(R&D) :在药物设计和开发阶段,应评估和监控潜在的遗传毒性杂质,确保其控制在安全摄入量范围内。质量保证(QA) :应根据ICH M7 R2 Addendum更新质量标准和检验流程,确保产品放行前符合规定要求。临床(Clinical) :在临床试验方案设计时,应考虑文件中的指导原则,特别是在涉及遗传毒性杂质的风险评估和管理方面。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保文件中关于leachables和extractables的控制措施得到实施,并监督相关质量风险管理流程。 RA(注册事务):在药品注册过程中,需要参考本文件准备和提交leachables和extractables的相关研究资料。 R&D(研发):在新药研发阶段,需依据本文件评估和控制leachables,确保药品的安全性和质量。 CMC(化学、制造和控制):负责执行leachables和extractables的具体研究工作,并确保数据的准确性和完整性。 工作建议:
QA:制定和更新内部SOP,以符合指南要求,并对相关流程进行审计,确保合规性。 RA:在注册文件中包含leachables和extractables的风险评估和控制策略,与监管机构沟通以获得批准。 R&D:在药物开发早期阶段进行leachables的风险评估,并选择合适的包装和制造组件以最小化风险。 CMC:进行leachables和extractables的实验研究,提供准确的数据支持药品安全性评估。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QC :必读。应熟悉药典文本的评估和推荐流程,确保质量控制标准与ICH区域药典要求一致。注册 :必读。了解药典文本在药品注册中的应用,确保注册文件符合ICH区域药典标准。研发 :必读。在药品研发阶段,需参考药典文本以确保产品符合ICH区域的药典要求。适用范围:
要点总结:
药典文本评估 :强调了对药典文本的系统评估,以确定其适用性和科学合理性。推荐流程 :明确了推荐药典文本的流程,包括专家评审和公开征求意见。协调一致 :鼓励ICH成员国在药典文本的采用上达成协调一致,以促进药品的国际流通。科学基础 :强调药典文本应基于科学证据,确保药品质量和安全性。更新与修订 :指出药典文本应定期更新和修订,以反映最新的科学进展和监管要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: 注册(Regulatory Affairs): 需熟悉平台数据上传流程及权限管理,提前准备CCI授权文件。 CMC(Chemistry, Manufacturing, and Controls): 针对Q13用例,梳理连续制造技术数据并制定共享策略。 临床(Clinical Development): 评估E19协议中选择性数据收集的合规性,确保SSDC方案符合平台讨论要求。 IT安全(IT Security): 验证企业内部系统与平台的安全兼容性,强化数据加密与访问控制。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必读。需更新质量管理体系,确保与GCP修订稿征求意见稿保持一致。 注册:必读。需掌握GCP修订稿征求意见稿内容,以便在注册申报中符合最新要求。 临床:必读。需根据修订稿调整临床试验操作流程,确保合规性。 研发:必读。需在药物研发阶段考虑GCP修订稿征求意见稿中对新技术应用的原则。 市场:必读。需了解GCP修订对市场准入和推广的影响。 适用范围:
文件概要:
以上仅为部分要点,请阅读原文,深入理解监管要求。