首页
>
资讯
>
MHRA 发布7份系列指南,支持全球首个去中心化制造监管框架
6月11日,英国药品和健康产品管理局(Medicines and Healthcare products Regulatory Agency,MHRA)宣布,为即将于7月23日生效的全球首个去中心化制造(Decentralized Manufacturing,DM,曾译为分散式制造,现参考我国官方对去中心化临床的翻译)框架,发布了七份指南文件,涵盖去中心化制造从临床、GMP到药物警戒的方方面面,为框架实施提供系统性支持,也为全球业界提供参考。
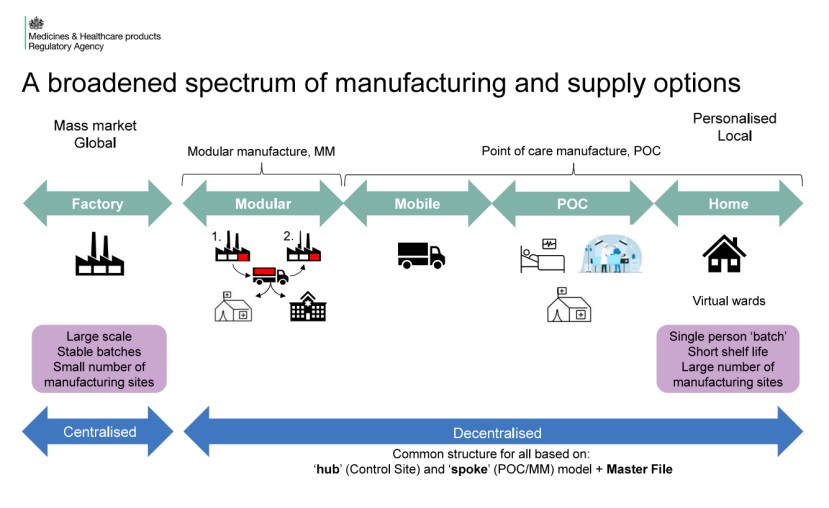
去中心化制造是一种新兴的药品生产模式,允许药品在患者使用地点附近或直接在使用地点进行生产。这种模式特别适用于那些货架期短、高度个性化或需要快速响应的药品,例如个性化癌症治疗药物和应急疫苗。与传统的集中式生产相比,去中心化制造能够提高药品生产的灵活性和效率,同时减少供应链风险,尤其是在应对紧急情况或小众市场需求时。
适用岗位:
- “注册”:必读。需确保所有药品标签符合英国法规要求,特别是未许可药品、研究用药品和药品的供应状态。
- “QA”:必读。负责监督标签的合规性,确保所有标签信息准确无误,特别是针对POC药品的即时使用要求。
- “研发”:必读。在研发阶段就需考虑标签要求,确保研发产品符合法规。
工作建议:
- “注册”:在药品注册过程中,特别关注标签的合规性,确保所有药品标签信息符合MHRA的最新指南。
- “QA”:在质量保证流程中,加强对标签的审查,确保无保留药品的标签符合即时使用的要求。
- “研发”:在研发阶段,提前规划标签设计,确保产品在上市前符合所有标签规定。
适用范围:
本文适用于在英国上市的所有药品类型,包括化学药、生物制品等,特别关注未许可药品、研究用药品和POC药品的标签要求。适用于Biotech、大型药企、跨国药企等各类企业。
文件要点总结:
本指南强调了药品标签在英国法规下的重要性,特别是针对未许可药品、研究用药品和药品供应状态的标签要求。特别指出,POC药品需要立即使用,制造商应预先在最终产品和/或转移容器上标注药品和患者标识符。对于POC药品,标签要求有所放宽,因为这些药品需要在制造后立即且完全使用,不会保留用于后续使用。此外,指南鼓励制造商对所有适用的最终产品和/或转移容器进行预标签,以确保药品和患者信息的准确性和可追溯性。这些要求旨在确保药品的安全性和有效性,同时满足监管合规性。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及建议:
- 注册(RA):必读。需熟悉去中心化生产(DM)的上市许可申请流程,包括资格、过程和要求,确保申请文件符合MHRA规定。
- 生产(Prod):必读。应确保生产许可证(MIA)符合DM要求,及时更新GMP合规证书,以避免MAA被拒。
- 质量控制(QC):必读。负责提供产品开发、生产和控制的一般原则,以及过程验证和/或扩展特性信息,以证明批次和/或生产地点之间的可比性。
适用范围:
本文适用于英国药品和保健品监管局(MHRA)发布的去中心化生产(DM)的化学药品上市许可申请,适用于Biotech、大型药企、跨国药企等企业类别。
文件要点总结:
MHRA发布的指南详细阐述了去中心化生产(DM)的上市许可申请(MAA)流程。申请DM需获得DM指定,确认符合法律测试。MAA应包含DM指定证据,并确认支持信息未变。若信息有重大变更,需重新申请DM指定。理想情况下,控制站点的生产许可证(MIA)应在提交时已适当调整以适应DM,若未完成,不影响MAA提交和验证。但若未能及时提供当前和适当范围的GMP合规证书,可能导致MAA被拒。产品开发、生产和控制的一般原则继续适用。可能需要过程验证和/或扩展特性信息以证明批次和/或生产地点之间的可比性,对于更复杂或潜在可变产品,所需数据量更大。若提出实时放行测试(RTRT)原则,应提供增强的产品和过程知识信息以支持此方法,并在P.3.5节提供代表性过程验证结果。RTRT策略应基于对制造过程和过程参数、中间控制与成品属性之间关系的深入理解。关于RTRT原则的更多信息可在实时放行测试指南和Eudralex第4卷附件17中找到。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
- PV(药物警戒人员):必读。需关注去中心化生产药品的制造变异性、产品可追溯性,以及如何整合这些因素到药物警戒活动中。
- QA(质量保证人员):必读。应确保质量管理体系支持药物警戒流程,特别是关于产品和批次信息的收集与报告。
- MAH(市场授权持有人):必读。需制定风险管理计划,包括对去中心化生产药品的特殊考虑,并确保产品信息及时更新。
文件适用范围:
本文适用于英国授权和未授权的药品(包括特殊药品和早期获取药品计划EAMS),特别针对去中心化生产(包括现场护理POC和模块化制造MM)的化学药品。发布机构为英国药品和保健品监管局(MHRA),适用于Biotech、大型药企、跨国药企等在英国授权的药品。
文件要点总结:
本文强调了去中心化生产药品在药物警戒方面的特殊挑战,尤其是在制造变异性和产品可追溯性方面。对于POC和MM药品,制造过程的质量与产品内在物质同等重要,制造步骤的微小变化可能影响产品质量及其安全性和有效性。因此,药物警戒活动必须考虑制造场所层面的差异,以及活性物质和产品层面的差异。此外,产品和批次的连续可追溯性是药物警戒的一个关键要求,以确保能够快速检测和评估新出现的产品特定安全问题。对于EAMS和特殊药品,本文提供了关于不良反应管理和报告、风险管理系统、定期报告以及特殊药品的风险管理计划的具体指导。这些要求旨在确保去中心化生产药品的安全性信息能够被有效收集和评估,以支持风险管理决策和患者安全。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
- 注册:必读。在提交CTA申请时,需确保包含DM(去中心化制造)指定的证据,并确认支持DM指定的信息未发生重大变化。如有变化,需重新申请DM指定。
- QA:必读。确保去中心化生产的IMP遵循GCP原则,包括产品开发、制造和控制的一般原则,以及IMPD的一般要求。
- 研发:必读。在提出RTRT(实时放行测试)原则时,需展示增强的产品和过程知识,以支持这一方法,并提供代表性过程验证结果。
适用范围:
本文适用于使用去中心化制造的临床试验药物(IMPs)的CTA申请和GCP,适用于MHRA监管下的化学和生物制品的临床试验,包括Biotech和大型药企。
文件要点总结:
本文强调了去中心化制造(DM)在临床试验中的应用,要求CTA申请中包含DM指定的证据,并确认支持信息未变。若信息有重大变化,需重新申请DM指定。对于使用DM IMPs的临床试验,必须有一个控制点,并在CTA申请中提出,该点需在提交CTA申请前获得控制DM所需模式的认证。此外,文件详细说明了产品开发、制造和控制的一般原则,以及IMPD的一般要求,包括批次分析数据和过程验证信息。对于采用RTRT原则的DM IMPs,需在IMPD中提供增强的产品和过程知识,以及代表性过程验证结果。最后,文件强调了在盲法试验中维护盲法完整性的重要性,包括制造和给药过程中的控制措施,以防止无意中透露治疗分配。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
- QA:必读。需理解去中心化生产(DM)的认定步骤,确保质量管理体系符合MHRA要求。
- 注册:必读。负责申请DM认定,需准备相关证据和数据支持申请。
- 研发:必读。在产品和工艺开发早期阶段,评估是否符合DM标准,及时与MHRA沟通。
适用范围:
本文适用于英国MHRA管辖下的化学药和生物制品,包括创新药和仿制药,原料药等。适用于Biotech、大型药企、跨国药企等各类企业。
文件要点总结:
MHRA发布的去中心化生产(DM)认定步骤指南,旨在评估申请组织提供的DM使用理由。DM分为现场制造(POC)和模块化制造(MM),需满足特定法律测试。申请DM认定应在产品和工艺开发早期进行,与MHRA早期沟通。认定成功后,MHRA会提供认定参考,用于未来监管提交。若支持信息发生重大变化,需重新申请DM认定。MHRA将在30天内初步决定,必要时60天内完成审查。认定结果不公开,有效期内有效。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
- “注册”:必读。需确保所有药品标签符合英国法规要求,特别是未许可药品、研究用药品和药品的供应状态。
- “QA”:必读。负责监督标签的合规性,确保所有标签信息准确无误,特别是针对POC药品的即时使用要求。
- “研发”:必读。在研发阶段就需考虑标签要求,确保研发产品符合法规。
工作建议:
- “注册”:在药品注册过程中,特别关注标签的合规性,确保所有药品标签信息符合MHRA的最新指南。
- “QA”:在质量保证流程中,加强对标签的审查,确保无保留药品的标签符合即时使用的要求。
- “研发”:在研发阶段,提前规划标签设计,确保产品在上市前符合所有标签规定。
适用范围:
本文适用于在英国上市的所有药品类型,包括化学药、生物制品等,特别关注未许可药品、研究用药品和POC药品的标签要求。适用于Biotech、大型药企、跨国药企等各类企业。
文件要点总结:
本指南强调了药品标签在英国法规下的重要性,特别是针对未许可药品、研究用药品和药品供应状态的标签要求。特别指出,POC药品需要立即使用,制造商应预先在最终产品和/或转移容器上标注药品和患者标识符。对于POC药品,标签要求有所放宽,因为这些药品需要在制造后立即且完全使用,不会保留用于后续使用。此外,指南鼓励制造商对所有适用的最终产品和/或转移容器进行预标签,以确保药品和患者信息的准确性和可追溯性。这些要求旨在确保药品的安全性和有效性,同时满足监管合规性。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
- QA(质量保证):必须熟悉新法规框架,确保生产过程和产品质量符合MHRA的新要求。
- 生产:需要了解模块化生产和床旁生产的具体规定,调整生产流程以符合新法规。
- 注册:负责更新和申请制造许可证,包括POC和MM产品的许可证变更快。
- 药物警戒:掌握新的不良反应报告要求,确保及时准确地向MHRA报告。
工作建议:
- QA:审查和更新质量管理体系,确保涵盖新法规中提到的所有要素,如POC和MM产品的特定要求。
- 生产:评估现有生产设施,确定是否需要升级或改造以适应新的生产模式。
- 注册:准备必要的文件和数据,以支持制造许可证的申请和变更。
- 药物警戒:更新药物警戒系统,确保能够收集和报告POC和MM产品的不良反应。
适用范围:
本文适用于人用药品,特别关注模块化生产(MM)和床旁生产(POC)的化学药品和生物制品。适用于英国MHRA监管框架下的Biotech、大型药企、跨国药企以及CRO和CDMO等企业。
文件要点总结:
MHRA发布的2025年人用药品模块化生产与床旁生产法规概述,强调了对创新药品的新监管框架,特别是那些货架期极短或需要高度个性化的产品。新框架要求在患者接受护理的地方(POC)或附近制造和供应药品,以确保药品的安全性、质量和有效性。POC药品可能涵盖多种类型,包括先进的治疗药品(ATMPs)、3D打印产品、血液制品和医疗气体。新法规详细定义了POC和MM相关术语,并要求持有制造许可证的公司必须遵守GMP原则,并维护必要的人员、场所和设备。此外,新法规还涉及了POC和MM产品的制造和组装义务、合格人员的责任、许可证变更、市场营销授权申请以及药物警戒要求。特别指出,POC和MM产品的制造必须在指定的控制场所进行,并且必须有相应的主文件(POC master file或MM master file)来详细描述制造或组装的安排。这些文件需要定期更新,并在MHRA要求时提供。对于药物警戒,新法规要求及时报告所有疑似不良反应,并与MHRA合作以识别和评估相关风险。
以上仅为部分要点,请阅读原文,深入理解监管要求。