首页
>
资讯
>
术语、算法和表格模板:FDA 审评新药安全性的工具
出自识林
术语、算法和表格模板:FDA 审评新药安全性的工具
2025-07-14
6月,FDA发布了两份政策和程序手册(MAPP),分别为MAPP 6025.8 《审评质量管理规范: OND 定制医学查询》 (Good Review Practices: OND Custom Medical Queries,OCMQs)和MAPP 6025.9 《审评质量管理规范:标准安全性表与图》 (Good Review Practices: Standard Safety Tables and Figures,ST&Fs)。这两份文件用于FDA内部药物审评,也可为药企的新药研发和申报注册工作提供指导。
这些工具基于FDA内部的生物医学信息与监管审评科学团队(Biomedical Informatics and Regulatory Review Science,BIRRS)的多年研究和实践经验。BIRRS页面上可以查阅具体的工具说明并下载配套指导性文件。
FDA并不要求申请人按照OCMQs术语和ST&Fs的格式提交申报资料,但基于对OCMQs和ST&Fs的充分了解,申请人可站在审评人员的角度对提交的安全性数据进行预先评估,识别风险,并提高沟通效率,其价值不言而喻。比起近日FDA大力推进的基于大语言模型(LLM)的AI工具Elsa ,这些基于传统信息技术和确定性算法的工具也许更能体现FDA审评的特点。
定制医学查询(OCMQs):可附带算法的标准术语集
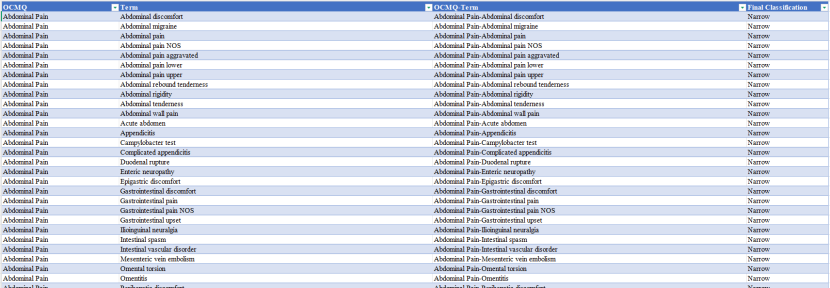
OCMQs是一套标准化的不良事件 (Adverse Event,AE)术语组合,专门设计用于审评人员在审查临床试验 AE数据时识别潜在的安全性问题。OCMQs通过将相关AE术语进行分组,提供了一种系统化的方法来检测安全性信号,从而提高审评效率和一致性。
通过标准化的术语分组,审评人员可以快速识别出可能的安全性问题,而无需逐一审查每个AE术语,不仅节省时间,还减少人为判断差异导致的不一致性。此外,OCMQs还支持算法组件,能够利用额外信息(如AE术语组合、实验室数据、合并用药 、病史或时间信息)识别更复杂的安全性信号。审评人员还可以根据具体的研究设计和安全性问题,对OCMQs进行调整和定制。
最新的OCMQs版本3.0基于MedDRA v26.0或更早版本的术语,可在FDA官网下载,未来将定期更新以反映最新的医学知识和技术进展。
药企也可以从中受益。通过熟悉OCMQs的术语分组和分析方法 ,药企可以在临床试验阶段提前识别和管理潜在的安全性问题。此外,OCMQs的标准化特性也有助于药企在申报注册过程中更清晰地向FDA展示其药品的安全性数据。
标准安全性表与图(ST&Fs):安全性数据展示模板
ST&Fs是一套标准化的临床安全性数据展示工具,包括综合指南(Integrated Guide,IG)和针对性分析指南(Targeted Analysis Guides,TAGs),用于审评人员高效、一致地识别和评估潜在安全性问题。
通过ST&Fs,审评人员能够更快速地生成安全性审查内容,并确保不同审评部门在产品安全性审查中的标准统一。ST&Fs的标准化表和图的设计可改善临床安全性数据的展示质量,可以更清晰地识别潜在的安全性信号,使审评过程更加高效和流畅。
ST&Fs的应用范围广泛,通常包括关键试验、注册试验,以及其他新药办公室(OND)临床审评人员关注的试验。这些试验可能在关键设计特征上与关键试验存在显著差异,例如导入期、富集设计、受试人群、持续时间或交叉设计 。此外,ST&Fs还可用于汇总分析,以提供更全面的安全性评估视角。
除了标准化的表和图,ST&Fs的针对性分析指南(TAGs)还用于进一步调查识别的安全性信号,或针对某些特别关注的不良事件(AESI)进行深入分析。FDA提供2份TAGs下载,分别针对肾损伤和肌肉损伤。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位:
注册(RA) :应熟悉OCMQs的使用和更新流程,以便在提交NDAs、BLAs及补充申请时,能够正确地应用OCMQs进行安全评估。临床(Clin) :在进行临床数据安全分析时,需了解OCMQs的标准分组,以识别潜在的安全问题。QA :监督OCMQs的开发、发布和政策,确保审评过程中的质量控制。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
本文件适用于“临床”(Clinical)岗位,因为他们需要评估临床安全数据并使用ST&Fs进行分析。 对于“统计”(Statistical)岗位,他们需要参与讨论分析方法,并进行定制分析以进一步评估潜在的安全信号。 “注册”(Registration)岗位也应阅读,因为他们负责提交新药申请(NDAs)和生物制品许可申请(BLAs)。 “QA”岗位应了解,以确保临床安全数据的展示和分析符合FDA的要求。 工作建议:
临床岗位:使用ST&Fs识别和评估潜在的安全发现,并在审查中呈现临床安全数据。 统计岗位:与临床团队合作,确定分析方法,并进行定制分析以进一步探索安全信号。 注册岗位:确保提交的申请符合ST&Fs的要求,并在必要时提供定制分析。 QA岗位:监督ST&Fs的使用,确保临床安全数据的质量和一致性。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。