|
首页
>
资讯
>
国内药政每周导读:附条件批准工作程序再征意见,参比目录第93批发布
出自识林
国内药政每周导读:附条件批准工作程序再征意见,参比目录第93批发布
2025-07-14
【药学研究】
7.7,【NMPA】关于发布仿制药参比制剂目录(第九十三批)的通告(2025年第24号)
截至目前,NMPA已发布参比制剂目录93批,CDE已公示95批。
识林用户可登录“中国参比制剂库”查阅。
【注册与变更】
7.7,【NMPA】再次公开征求《药品附条件批准上市申请审评审批工作程序(试行)(修订稿征求意见稿)》及政策解读意见
上次征求意见是2023年8月,现行版本是2020年7月。
相比2023年版本,改动较大,列举比较重要的几点:
- 删去了经技术审评发现不满足附条件批准上市要求的,“申请人对审评结论有异议的,可以按照药品注册审评结论异议解决的有关程序提出。药品注册申请审批结束后,申请人对行政许可决定有异议的,可以依法提起行政复议或者行政诉讼。”
- 申请延期的补充申请原则上不超过一次。且“注册证书有效期保持不变,有效期届满后,持有人应当暂停销售。”
- 新增“如完成的临床研究同时可支持新增适应症,持有人可提出新增适应症上市申请,在该申请中同时提出附条件批准的适应症转为常规批准。”
- 新增“附条件批准上市的药品,拟发生持有人变更的,受让方应当充分评估药品安全有效性研究情况,确保前期研究完整转移,确保后续按期完成确证研究。受让方在提交持有人变更申请前,应就研究进展、下一步研究计划等与药审中心进行沟通交流,沟通交流同意的,方可提出持有人变更申请。”
【新药批准和报产】
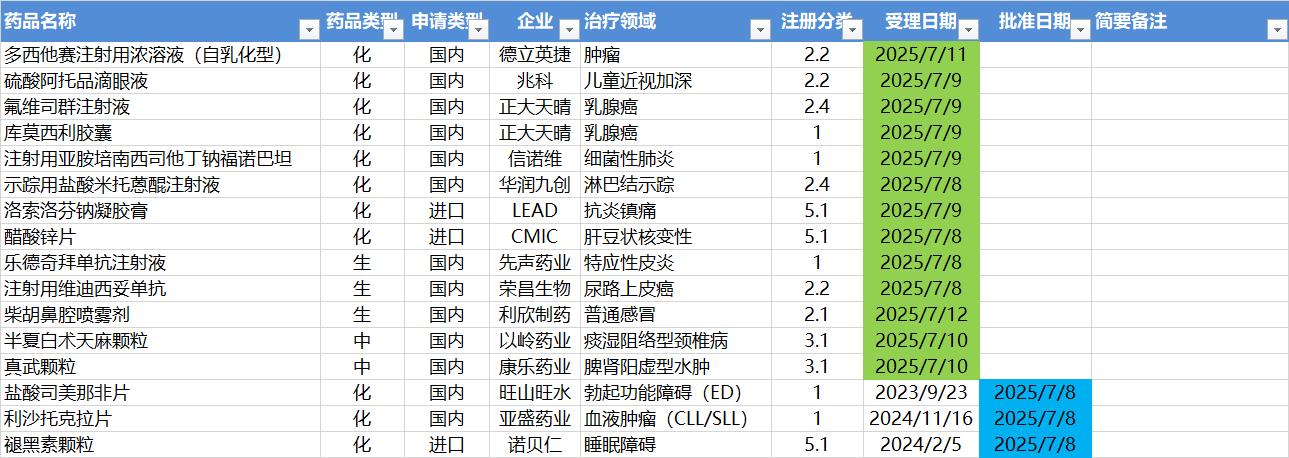
7.7-7.13,NMPA发布3个新药批准,CDE受理13个新药上市申请
注:仅列出新药(包括改良型新药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林®版权所有,未经许可不得转载
适用岗位: - 注册(RA):必读。需关注新发布的参比制剂目录,以便在仿制药注册申报时准确引用。
- 研发(R&D):必读。需根据参比制剂目录调整研发策略,确保产品质量与疗效。
- 质量保证(QA):必读。需确保生产过程和产品质量符合参比制剂标准。
适用范围:
本文适用于化学药品的仿制药,包括创新药、仿制药、生物类似药及原料药等,由NMPA发布,适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。 文件要点总结:
国家药监局发布了第九十二批仿制药参比制剂目录,旨在进一步明确仿制药研发和评价的参照标准。本批目录包含了多个未在国内上市的原研药品,以及部分在国内上市的原研药品和原研地产化药品。特别指出,这些参比制剂来源于美国橙皮书、欧盟上市药品、日本上市药品等,显示了国际药品监管机构之间的互认和合作。目录中还特别提到了增加变更后上市许可持有人的情况,这要求企业在注册和生产过程中密切关注许可持有人的变更,确保合规性。这些参比制剂的发布,对于指导仿制药企业在研发、注册和生产过程中确保产品质量和疗效的一致性具有重要意义。 以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位及工作建议: - RA(注册):必读。需熟悉修订后的医疗器械分类规则,以便在注册申报时准确分类医疗器械产品,确保合规性。
- QA(质量管理):必读。应根据新规则更新质量管理体系,确保产品分类与监管要求一致。
- R&D(研发):必读。在研发阶段即考虑医疗器械的分类,以指导产品设计和开发。
适用范围:
本文适用于中国境内所有医疗器械的分类工作,包括化学药、生物制品、疫苗和中药等类型的医疗器械,涉及创新药、仿制药、生物类似药、原料药等注册分类,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结:
《医疗器械分类规则(修订草案征求意见稿)》旨在规范医疗器械分类,明确了医疗器械按照风险程度分为三类,并详细定义了相关术语。修订内容主要包括对医疗器械分类目录的动态调整、医疗器械附件的分类原则、以及对医用敷料等特定产品的分类管理。特别强调了侵入器械、重复使用手术器械、植入器械等的定义更新,以及对医疗器械分类判定表的调整。新增了对某些特定医疗器械的管理类别规定,如可吸收医疗器械、医用敷料等。此外,明确了国家药品监督管理局在医疗器械分类规则起草、分类界定指导原则和分类目录制定中的职责,并废止了2015年的旧版规则。 以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - 注册(RA):必读。需熟悉附条件批准的申请流程和要求,以便在药物临床试验期间及时与药审中心沟通,准备和提交申请资料。
- 研发(R&D):必读。需了解附条件批准的技术要求,确保临床研究计划和关键临床试验设计符合规定。
- 临床(Clin):必读。需掌握附条件批准的临床研究计划和上市后研究要求,以确保临床试验的顺利进行和数据的有效性。
适用范围:
本文适用于在中国注册的化学药、生物制品、疫苗和中药,包括创新药、仿制药、生物类似药和原料药。适用于Biotech、大型药企、跨国药企及CRO和CDMO等企业类别。 文件要点总结:
国家药品监督管理局制定了药品附条件批准上市申请审评审批工作程序,旨在加快具有临床价值的急需药品上市。适用条件包括公共卫生急需药品和重大突发公共卫生事件急需疫苗。工作程序涉及早期沟通、上市前沟通、提交申请、审评审批及上市后要求。鼓励申请人在临床试验期间与药审中心沟通,以确保附条件批准的临床研究计划和关键临床试验设计符合要求。审评通过后,将发给药品注册证书,并明确有效期、上市后研究工作及完成时限。上市后,持有人需采取风险管理措施,并在规定期限内完成相关研究。若研究证明药品获益大于风险,将换发有效期为5年的药品注册证书;否则,将注销注册证书。工作要求强调沟通交流、申报材料的合规性及技术审评的具体要求。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |