|
首页
>
资讯
>
读 CDE《2025年度药品审评报告》
出自识林
读 CDE《2025年度药品审评报告》
2026-05-19
2026年5月13日,CDE发布《2025年度药品审评报告》,大量数据显示我国药监和药企收获颇丰。
按照我国药品注册分类相关标准,全年批准上市 1 类创新药 76 个品种,其中新机制新靶点药物 11 个,26 个品种(34.2%)通过优先审评审批程序批准上市,15 个品种(19.7%)附条件批准上市,15 个品种(19.7%)在临床试验期间纳入了突破性治疗药物程序。
全年批准罕见病用药 48 个品种(未包括化学药品 4 类仿制药),其中 12 个品种(24.5%)通过优先审评审批程序加快上市;批准儿童用药 138 个品种,包含 98 个上市许可申请,其中25 个品种(18.1%)通过优先审评审批程序加快上市;另批准 40 个品种扩展儿童适应症;批准放射性药品 6 个品种,其中 3 个品种(50.0%)通过优先审评审批程序加快上市;批准境外已上市境内未上市的药品(化学药品 5.1 类、治疗用生物制品 3.1 类和预防用生物制品 3.1 类)80 个品种,其中 57 个为首次批准上市,23 个为已上市药品增加适应症。
以下内容并非严谨研究,数据的异步对比也难以得出可靠结论,仅从不同角度作简单导读,供读者参考,欢迎指正。
对比欧美数据:数量已超越,“质量”和加速效率仍待努力
美国 FDA 于 2026 年 1 月 23 日发布了其 2025 年度 FDA 新药审批总结报告。报告显示药品审评与研究中心(CDER)的46 个获批新药中,20个(43%)被确认为首创(first-in-class),23个(50%)被认定为孤儿药,18 个(39%)获得快速通道认定,15个(33%)被认定为突破性治疗药物,21个(46%)获得优先审评认定,11个(24%)获得加速审批。CDER 对 2025 年批准的所有新药中的 33 个(72%)使用了一项或多项加快开发和审评方法。另外,39 个(85%)首轮获批,32 个(70%)先于任何其它国家在美国获批。生物制品审评与研究中心(CBER)批准了12个新生物制品。
1月15日,EMA发布了《2025年的人用药》报告,列出了EMA全年出具的104份批准意见,包含38种新活性物质、16种孤儿药、4种先进治疗药品(ATMP)、41种生物类似药、10种仿制药,以及3款通过加速审评、6款通过PRIME计划、8款通过附条件批准及2款通过特殊情况批准等特殊途径上市的药品。
对比可见,中国创新药获批数量已超过美国,但美国批准更多首创新药、更多利用加速通道、高达70%的全球首发上市,显示出审评效率和全球引领能力仍明显领先。欧洲则相对滞后,不仅新分子数量较少,加速与特殊通道利用也有限,与中美已不在同一量级。
看“受理”和“审结”:“一进一出”中的资源和效率平衡把握
报告包含大量数据,且经过较为充分的整理,包含历年对比和相应图表,已经相当直观。本文尝试从另一个角度,即从不同维度的“受理”和“审结”数据(“一进一出”)来看我国药监对工作量和工作效率的把握,这也直接关系到药企获批速度。
看总量:双双迈向“两万”时代,CDE量入为出
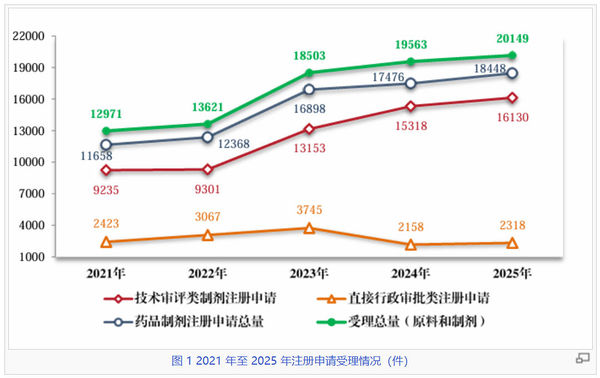
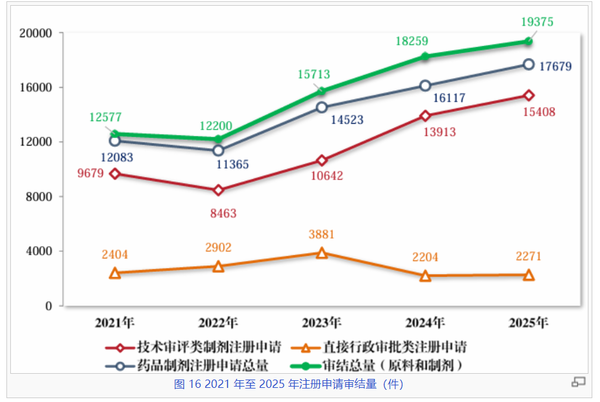
在政策引导与产业创新的双重驱动下,我国药品受理与审结数量自2021年起稳步攀升,至2025年双双迈向“两万件”大关。数据显示,2025年受理总量(含原料和制剂)达到20149件,较2021年的12971件大幅增长约55.3%;与此同时,审结总量也由2021年的12577件攀升至19375件,增幅达54.1%,呈现高位运行、动态平衡的态势,反映出审评部门在面对日益增长的申报压力时,尽力通过优化流程与科学监管,维持审评效能的同步释放。
从结构上看,技术审评类制剂申请是绝对主力,占据了制剂申请总量的近九成。2025年,技术审评类制剂受理量达到16130件,审结量达到15408件,其增长曲线与总量趋势高度吻合。尽管2022年曾出现过小幅波动,但自2023年起,无论是受理还是审结均同步上升。与此同时行政审批类申请在2023年达到阶段性峰值后逐步回归常态,有望腾出资源向技术审评倾斜。
在保持高强度审评产出的同时,多年来审结量与受理量之间较小的差距预示着申报积压问题得到了有效控制(尽管这是异步数据)。站在2026年的“十五五”新起点上,这份成绩单不仅印证了中国制药产业过去10年的成就,也为全球医药创新力量进入中国市场提供了更为可靠的准入预期。
看药品类型:化药增速放缓,生物制品和中药稳步提升
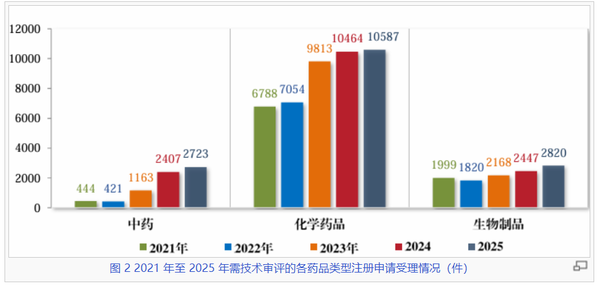
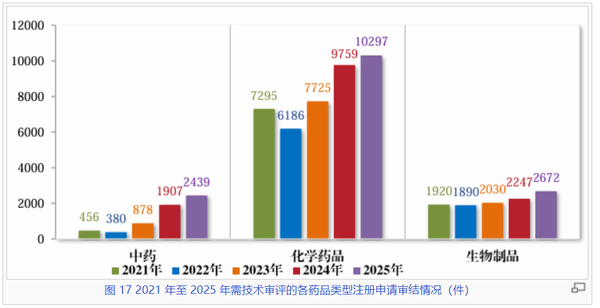
在2021至2025年的审评周期内,化学药品始终是审评工作的核心板块,以超过65%的份额继续引领。其2025年的受理量与审结量双双突破万件大关(受理10587件,审结10297件),其中化药板块的审结量较2022年的低谷(6186件)反弹了近66%,仍是行业坚实基础。
生物制品领域则展现出了韧性与成长潜力。2025年受理量达到2820件,较2021年增长约41%,其审结量也同步提升至2672件。作为前沿生物技术转化最为集中的领域,生物制品保持了持续向上的增长曲线,且受理与审结相当匹配度,显示出该板块研发与监管衔接的成熟度不断提升。
五年间,中药受理量从2021年的444件飙升至2025年的2723件,增幅高达513%。这种迅猛增长一定程度体现了国家促进中医药传承创新发展政策的深远影响。2025年审结量也保持了2439件的高效产出。
看注册分类:创新药持续收获,仿制药现拐点,补充申请高企
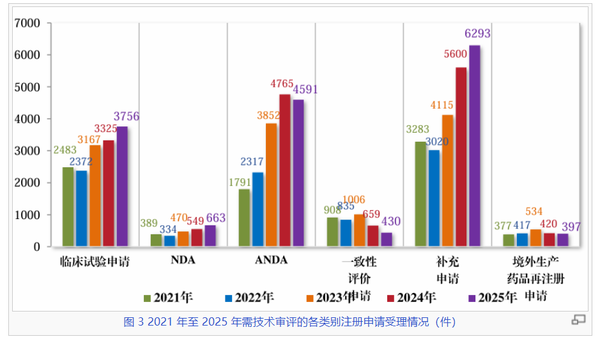
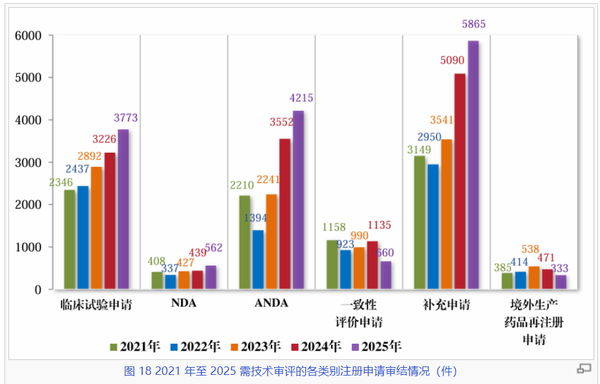
在技术审评的细分数据中,临床试验申请(IND)与新药上市许可申请(NDA)的双增长是最具行业风向标意义的信号。2025年临床试验申请受理量达到3756件,较2021年增长约51.3%,我国医药创新的“蓄水池”依然充盈。NDA受理量从2021年的389件稳步攀升至2025年的663件,审结量也同步达到562件的历史新高,标志着中国创新药研发已进入收获期,且可期待未来仍将保持。
仿制药(ANDA)板块,ANDA受理量在2024年达到4765件的高峰后,2025年略微下滑至4591件,“拐点”已现;而审结量则在2025年继续大幅增长,达到4215件,结合一致性评价申请呈现出逐步回落的态势,2025年受理量降至430件,已大幅低于审结量660——这可能预示着我国存量仿制药的开发和一致性评价放缓。
与此同时,补充申请在2025年受理量飙升至6293件,审结量达到5865件,稳居所有类别之首。这体现了已上市药品在工艺优化、规格变更及增加适应症方面的活跃需求,但也体现着相当的审评压力。
看三大加快机制:肿瘤药占比最高,监管调控审评资源
加快上市注册程序是鼓励药物创新、满足临床急需的重要制度安排。2025年,我国通过突破性治疗药物认定、附条件批准、优先审评审批等多元通道,持续优化审评资源配置。
突破性治疗药物:纳入多,拒绝纳入的更多
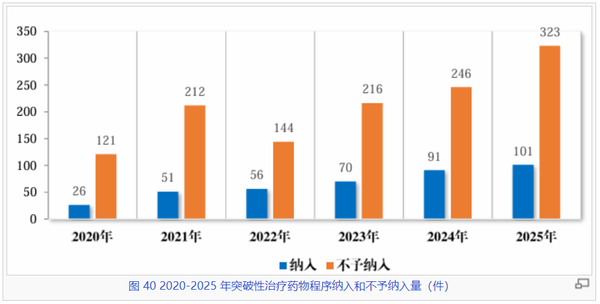
作为中国药品审评审批制度改革的重要一环,突破性治疗药物程序已成为创新药研发的“快车道”,也可谓是一根引导产业向原始创新靠拢的指挥棒,更能为患者带来优效新药可及的曙光。
数据显示,2020年至2025年间,该程序的纳入申请数量呈现显著增长态势。2025年,“纳入”数量首次突破百件大关,达到101件,较2020年的26件翻了近四倍。不过“不予纳入”的数量增速更快(2025年达到323件),这恰反映了监管部门对突破性标准的严格把控,确保真正具有临床优势的潜力新药脱颖而出。
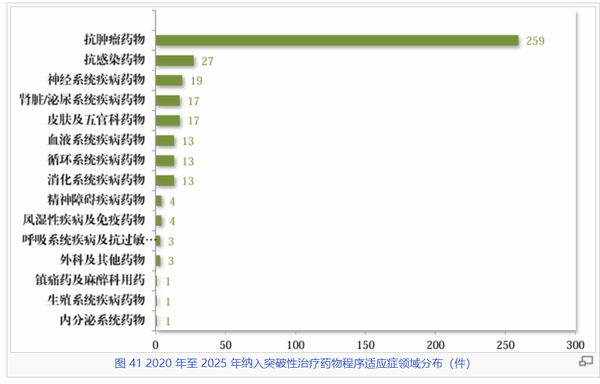
从突破性治疗药物的治疗领域分布来看,抗肿瘤药物展现出压倒性的研发活力,以259件的绝对数量稳居榜首。这显示出药企在攻克癌症这一世界性难题上的高度集中和持续投入。紧随其后的是抗感染药物(27件)、神经系统疾病药物(19件)以及肾脏/泌尿系统疾病药物(17件)。
附条件批准:进入平台期,存量陆续“转正”
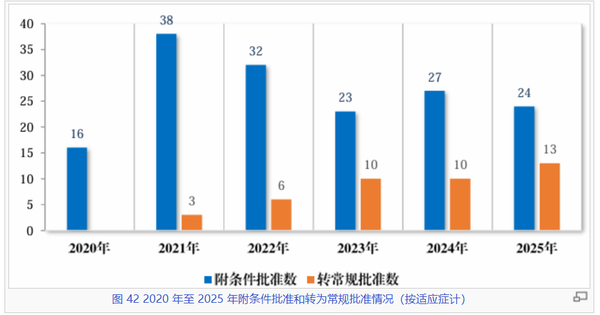
作为缩短重疾患者获药时间的重要监管手段,附条件批准制度在过去五年间发挥了关键作用。数据显示,附条件批准数量在2021年达到38件的高峰后,近年来维持在每年20-30件的区间稳步运行。2025年,共有24件药品或适应症通过该路径获批上市。这一趋势的变化反映了监管部门对附条件批准适用范围的把握导向,即不再单纯追求“快”,而是更加强调不可替代的临床价值,确保每一款通过该路径上市的药物都能精准解决现有的临床治疗空白。
与此同时,“转常规批准”数据的显著上升。2025年“转正”品种数量达到13件,创下新高。附条件批准绝非“终点”,企业在产品获批后必须履行承诺,按时完成上市后研究,形成“及时转正,有进有出”的动态管理。
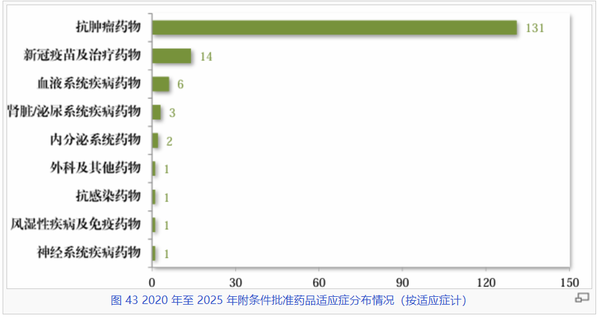
在疾病领域分布上,抗肿瘤药物以131件的累积数量依然占据绝对优势(排名第二的14个新冠药更多是应急使然),这一定程度凸显了该程序在攻克恶性肿瘤等重大疾病方面的政策导向作用。
优先审评审批:CDE自主灵活分配有限资源
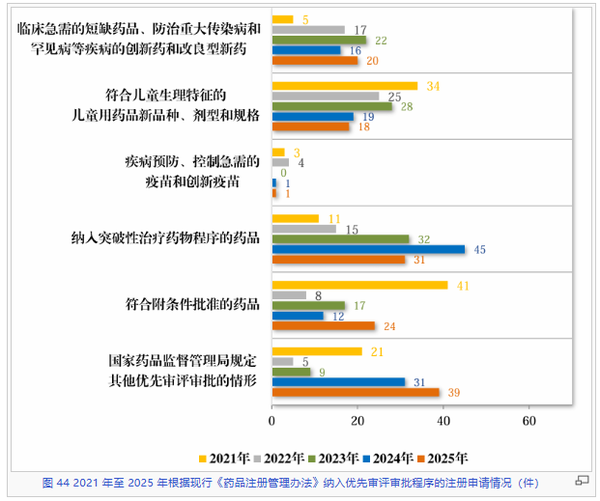
优先审评审批程序作为我国缩短药品审评周期的核心制度,在2025年展现出了更为均衡且精准的态势。从入选的情形来看,“临床急需、罕见病及重大传染病”相关药品的优先审评数量稳步回升。“符合附条件批准的药品”在经历了三年的低位运行后,于2025年显著反弹至24件。儿童用药在2025年入选优先审评的品种数量(18件)较往年高峰有所回落。
相比之下,“国家药监局规定的其他优先情形”在2025年增至39件,占比最高,反映出监管部门在调配审评资源时掌握主动。
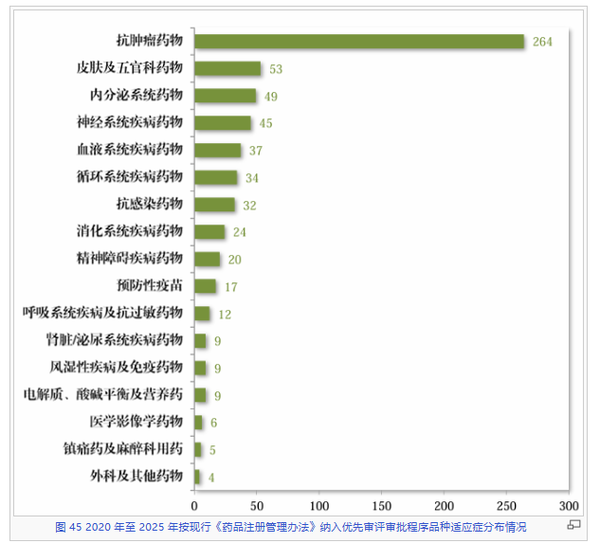
从治疗领域累积分布来看,又是抗肿瘤药物以264件的压倒性优势,成为优先审评程序最大的受益板块。紧随其后的是皮肤及五官科药物(53件)、内分泌系统药物(49件)以及神经系统疾病药物(45件)。
值得注意的是,尽管肿瘤药又是遥遥领先,但与其他领域药物的数量差距小于上述他两大机制,这也可能从侧面体现出监管部门倾向于自主灵活把握审评资源,而非全然跟从市场热点。
作者:识林-实木
识林®版权所有,未经许可不得转载。
【文件概要】
该报告总结了FDA药品评价与研究中心(CDER)2025年批准的46种新药(含34种新分子实体和12种生物制品),其中50%针对罕见病(23种获孤儿药资格),70%在美国首次获批,72%通过快速审评程序(含快速通道、突破性疗法、优先审评或加速批准)。创新药涵盖肿瘤、罕见病、非阿片类镇痛药等领域,包括首款Barth综合征疗法(Forzinity)和首款非阿片类镇痛药(Journavx)。CDER全年共批准123项产品(含新剂型),18种生物类似药(4种针对新参照产品),并首次公开19份完整回应函(CR letters)以提升透明度。审评效率方面,96%新药符合PDUFA目标时限,85%实现首轮批准。 【适用范围】
本文适用于美国市场的新药(化学药、生物制品)及生物类似药开发企业,包括创新药企、生物技术公司及跨国药企,尤其关注罕见病、肿瘤、感染性疾病等领域。涉及监管机构为FDA,涵盖NDA/BLA申请流程及加速审批路径(如突破性疗法、优先审评)。 【影响评估】
本文显示FDA持续优化审评效率,加速创新药上市,尤其利好罕见病和未满足临床需求领域。生物类似药审批增加可能加剧市场竞争并降低药价。公开CR函将促使企业更早优化申报策略,但需应对更高的透明度要求。 【实施建议】 - 注册:必读。需关注加速审批路径的适用标准及CR函公开后的数据要求调整。
- 临床:必读。重点分析罕见病和突破性疗法的试验设计,优化与FDA的早期沟通。
- 研发:必读。优先开发首创新药(如非阿片类镇痛药)及罕见病适应症,利用孤儿药政策。
- 市场:关注生物类似药竞争格局及新剂型产品的差异化策略。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |












