首页
>
资讯
>
近期重点483和警告信概要,识林 AI 为您“读缺陷”
出自识林
近期重点483和警告信概要,识林 AI 为您“读缺陷”
2026-01-17
识林目前收录4种检查报告,包括FDA警告信(药品警告信提供人工翻译)和483,欧盟缺陷信,以及WHO的公共检查报告。正在规划收录日本PMDA的“橙色警告信”。
此外,识林还将警告信和483分别整理成数据库,从国别、产品类型、检查官、缺陷关键词、法规条款等多个维度进行拆解。
一个新的变化是,识林将会员们喜闻乐见的AI 导读引入了483和警告信,为大家先“读缺陷”,高效抓取关键信息。详见文末。
*识林免费用户可查阅警告信等检查报告原文,会员可查阅双语版本和“警告信数据库” 、“483数据库” 。
*点击“帮助中心” 获得更多识林使用技巧。
近期更新的重点483和警告信及其缺陷概要
*均基于识林AI导读内容编辑,仅供参考。
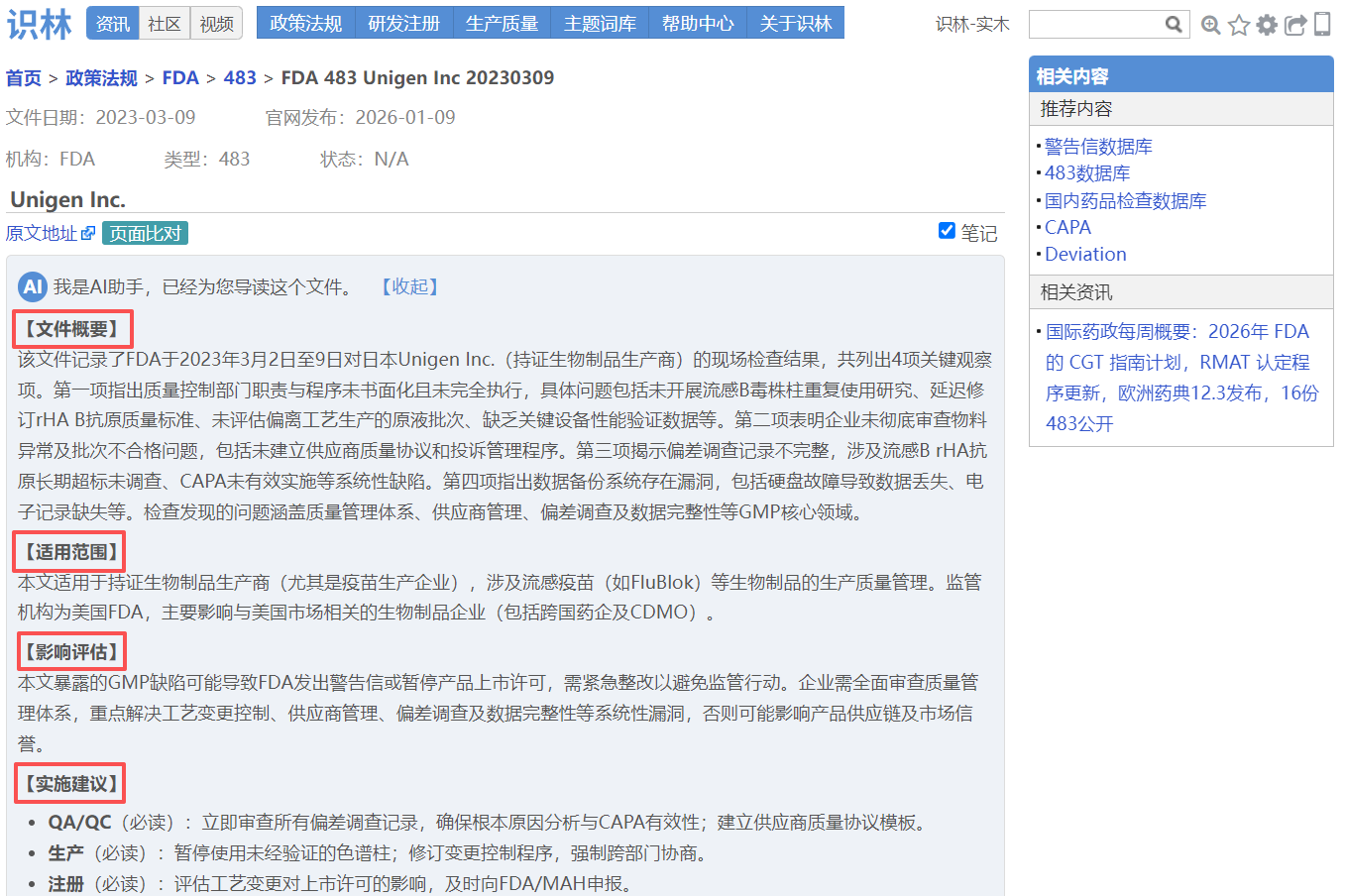
【483】美国 Aurobindo Pharma USA Inc
关键词: 印度药企公司总部,供应链安全,处方药盗窃事件
该483记录了FDA对Aurobindo Pharma USA Inc公司总部进行的现场检查结果,指出其未能建立符合《药品供应链安全法案》 (DSCSA)要求的验证 体系。检查发现两项主要观察项:第一,企业现有程序(SOP-ABN-QA-0040和SOP-ABN-DC-0004)未涵盖可疑产品识别、与贸易伙伴协同调查、非法产品判定及通知FDA的完整流程,且缺乏对高风险非法产品的24小时报告机制和产品标识验证响应流程;第二,企业在2023年5月发生处方药货物盗窃事件后,未按法规要求在24小时内向FDA提交非法产品通知。文件强调这些观察项仅为检查发现,不构成最终合规结论,企业需提交整改计划或异议说明。
【483】法国 Fareva Amboise
关键词: 无菌保证,气流模式,培养基灌装 ,清洁
该483记录了FDA对Fareva Amboise无菌 生产设施的检查结果,揭示其在无菌保障体系中的系统性缺陷 。文件指出无菌工艺 验证未充分评估气流模式(如A级区湍流、干预操作导致污染 风险),且培养基灌装设计未模拟商业生产的干预频率。环境监测 程序存在漏洞,包括未对A级区操作人员实施充分监控、消毒程序缺乏科学验证。设备管理方面,清洁验证 未覆盖最难清洁部位,非专用设备存在交叉污染 风险。数据可靠性缺陷涉及计算机系统 无审计追踪 、人工修改测试参数。质量管理体系 未能确保批记录 完整性(如缺失干预记录),且目检程序未建立有效缺陷检测标准。
【483】印度 Immacule Lifesciences Private Limited
关键词: 质量体系,灌装线设计,烟雾试验 ,括号法 ,计算机化系统权限
该483记录了2025年9月FDA对Immacule Lifesciences无菌制剂生产设施的检查结果,共提出8项关键缺陷。文件指出质量体系存在系统性失效,包括质量控制 部门未书面定义职责、环境监测趋势分析不充分、无菌操作程序执行偏差(如层流罩消毒 不当与物料 转移污染风险)。灌装线设计缺陷导致过度人员干预,且烟雾试验未真实模拟商业化生产条件。环境监测布点缺乏科学依据,沉降碟位置被物料遮挡。无菌工艺验证采用“括号法”但未评估所有污染风险,最终灭菌 产品的干预未纳入培养基灌装设计。计算机化系统(LIMS/SCADA)权限管控不足,存在数据篡改风险。实验室调查未充分模拟根本原因 ,部分批次放行未考虑稳定性数据。人员培训体系缺陷体现为目检 工具包七年未更新,操作员资质评估未覆盖最差产品条件。
【483】印度 Hetero Labs Limited, Unit-IX
关键词: API ,“影子”QC,“影子”仓库,原始记录撕毁
该483记录了印度Hetero Labs Limited第九生产单元在2025年9月检查中的6项关键缺陷。报告指出质量控制部门未书面化职责,且违规使用未注册实验室检测拟销往美国的原料药 ,该实验室未在DMF中披露。检查发现未注册实验室存有与主工厂批号匹配的原料药样品、中间体 及未受控检测记录,但工厂无样品转移文件或质量协议 。第二项缺陷涉及原料药及中间体在无接收、存储记录的情况下放行至无证仓库,缺失温度监控和隔离措施。第三项缺陷揭示批生产记录 中原料用量与实际产量不符,仓库中存有未记录的额外批次。第四项指出质量部门未有效管控GMP 文件,包括原始记录被撕毁及签名造假。第五项缺陷为标签 发放与使用未核销,导致批次追溯困难。最后一项为未调查未注册实验室检测的超标 结果,仅记录合格数据。
【483】中国台湾 UBI Pharma Inc.
关键词: AQL,烟雾试验 ,FAR,变更 ,偏差调查
该483记录了FDA对位于中国台湾的UBI Pharma无菌注射剂 生产的检查结果,指出五项关键缺陷。第一,目检设备未针对特定产品缺陷完成确认,缺陷样品库管理不规范,导致AQL检测失效(21 CFR 211.68 )。第二,无菌控制程序存在漏洞,包括烟雾研究设计缺陷、操作员干预不当及层流 中断处置缺失(21 CFR 211.113 )。第三,质量协议过期导致FAR未及时提交,投诉调查不充分(21 CFR 211.198 )。第四,变更控制未执行,CAPA 有效性不足(21 CFR 211.100 )。第五,偏差调查未覆盖已分销批次(21 CFR 211.192 )。文件强调企业需系统性整改设备确认 、无菌保障及质量体系。
【警告信】美国 Ralph A. DeFronzo, M.D./University of Texas Health Science Center at San Antonio
关键词: 临床研究者,临床方案遵循,IRB,违规入组
该警告信指出临床研究者在执行两项糖尿病药物临床试验 时违反21 CFR 312 法规,具体涉及未遵循研究方案和未及时向IRB报告变更。第一项违规为入组7名不符合血压、eGFR或禁用药物(SGLT2抑制剂/GLP-1受体激动剂)标准的受试者 ,研究者 虽事后修改方案并获IRB批准,但滞后超过一年,且纠正措施未充分说明未来如何确保监督。第二项违规为未及时报告研究设计从双盲 随机 试验变更为包含预试验的两阶段研究,导致所有受试者均接受试验药物 且知情同意文件 未同步更新。FDA强调研究者需确保方案依从性以保护受试者权益和数据可靠性 ,并要求15个工作日内提交详细整改计划。
【警告信】加拿大 Apotex Inc.
关键词: 泄漏测试,偏差调查,隔离器清洁维护,颗粒污染
该警告信指出Apotex公司无菌药品生产存在三项重大CGMP违规。第一,偏差调查系统失效,包括未及时调查泄漏测试失败、容器密封性缺陷及稳定性 样品问题,且未评估对已分销批次的影响。第二,无菌控制不足,表现为隔离器清洁维护缺陷、手套破损应急处理不当、环境监测设计不合理。第三,设备管理失当,如颗粒物污染未及时解决,升级延迟。FDA要求企业提供独立评估报告及整改计划,涵盖偏差调查系统、CAPA程序、变更管理 及无菌操作风险分析。企业已召回 部分产品并暂停生产,但需系统性改进质量体系,包括加强质量部门权限及管理层监督。
【警告信】美国 Liebel-Flarsheim Company LLC
关键词: 生物负荷,内毒素 OOS,洁净区 维护,制水系统
该警告信基于2025年FDA对Liebel-Flarsheim公司的现场检查,指出其违反21 CFR 210 /211 的CGMP要求,具体涉及两大核心问题。第一,企业未彻底调查40余起生物负荷超标(含TNTC结果)及内毒素OOS事件,调查结论缺乏科学依据,CAPA措施延迟至2026年完成,且过度依赖最终灭菌 工艺而忽视过程控制风险。第二,设施设备存在系统性缺陷,包括洁净区维护不足(HEPA密封胶带修补、设备锈蚀)、制药用水 系统腐蚀泄漏,且环境监测长期显示微生物污染 ,但整改计划延至2027年。FDA要求企业提交独立回顾性评估报告,涵盖所有无效化OOS结果的实验室与生产根本原因 分析,并全面整改微生物控制体系、设施设计及水系统。监管措施包括可能扣留出口证书、暂停新申请审批及启动产品召回。
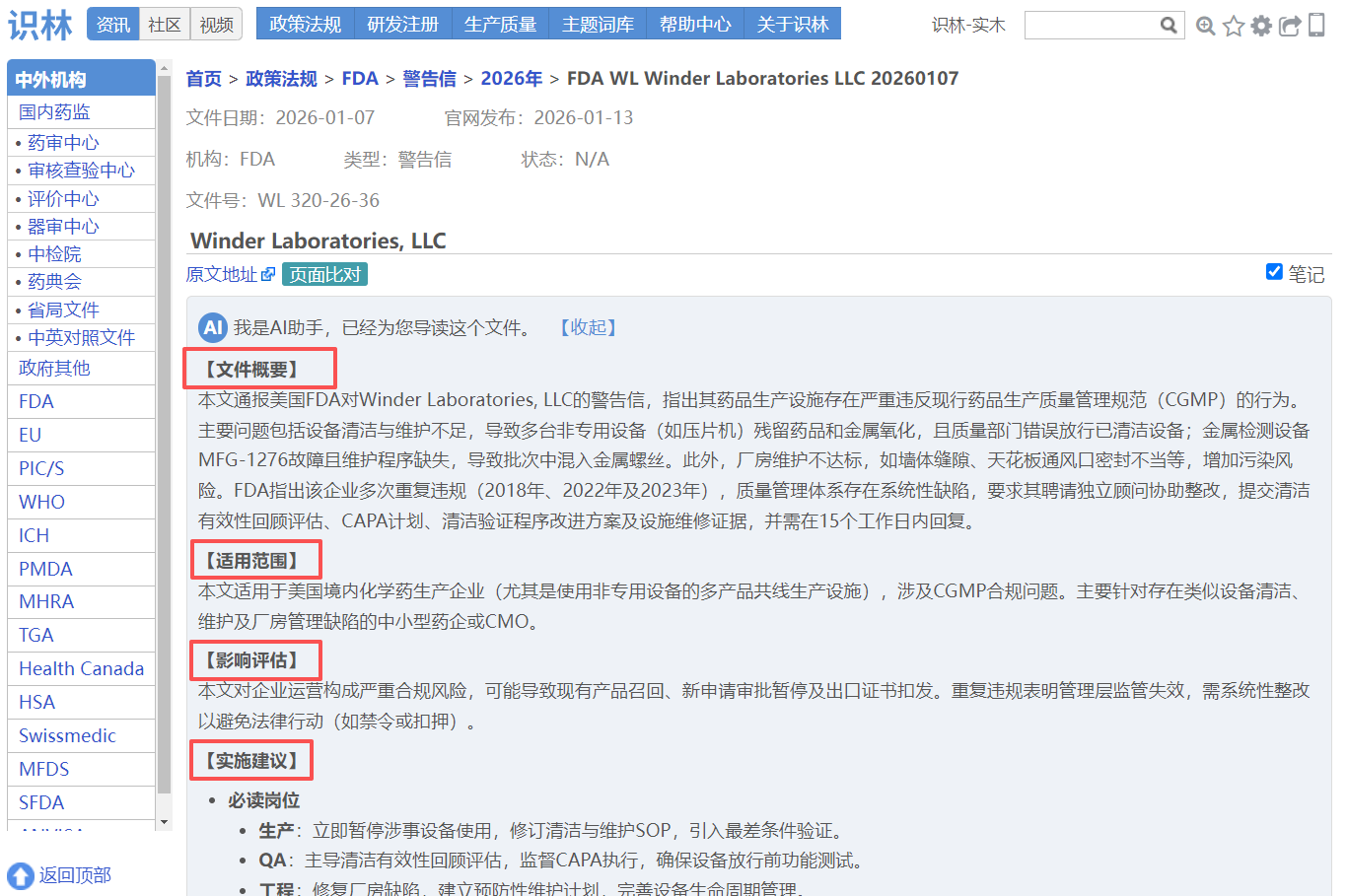
【警告信】美国 Rhyz Analytical Labs
关键词: 外包QC实验室,微生物OOS调查,CAPA有效性
该警告信指出受检实验室存在两项关键CGMP违规:未彻底调查微生物检测OOS结果(21 CFR 211.192 ),以及质量部门未履行书面程序及监督职责(21 CFR 211.22(d) )。具体问题包括多次以“未知实验室错误”草率结案,未科学论证污染源即依赖复测数据,且CAPA未评估有效性;质量体系缺乏偏差管理、变更控制等核心程序。FDA要求提供三年内OOS 结果的独立回顾性分析报告,并提交质量体系全面评估及整改计划,涵盖QU职能强化、实验室趋势监控及调查流程修订。文件援引FDA质量体系指南,强调需通过风险管理 实现CGMP合规。
【警告信】美国 Catalent Indiana, LLC
关键词: 动物毛发污染,关键无菌设备验证,环境监测方法
该警告信指出Catalent Indiana工厂在CGMP合规性上的系统性缺陷,主要涉及偏差调查 不足和无菌控制失效。具体问题包括对胶塞 携带哺乳动物毛发污染的调查不充分,未及时追溯污染源至供应商,且未有效实施CAPA ;无菌工艺 中未验证关键设备(如胶塞安装部件)的清洁效果,环境监测 方法(接触皿替代拭子)不足以检测微生物污染。FDA要求企业提交独立评估报告,涵盖偏差调查体系全面整改、供应商质量协议修订、无菌工艺再验证 ,以及污染 风险的全设施评估。文件强调需回溯审查客户投诉数据,并制定数据驱动的持续改进计划。
【警告信】美国 Darmerica, LLC
关键词: 原料药 ,供应商管理 ,质量投诉,放行检测,标签误导
该警告信指出Darmerica, LLC在API生产中的多项CGMP违规行为,包括质量体系失效、供应商管理缺陷及标签错误。文件首先概述了企业因质量部门(QU) 未履行监管职责导致API掺假的违法事实,具体表现为供应商评估不足(如接受虚假检查声明)、未调查质量投诉(如未追溯污染批次的其他客户)及放行前检测缺失(如提前分销未完成分析的API)。其次,文件详述了标签误导问题(如隐瞒GLP-1 API真实名称)和注册违规(如未正确提交510(j) 清单)。FDA要求企业在15个工作日内提交系统性整改计划,涵盖供应商重审、客户通知、QU职能强化及第三方审计。
将AI导读接入483和警告信
自从AI导读接入法规指南以来,得到了许多会员的积极反馈。警告信和483包含大量文字内容,483又往往是扫描件pdf的格式,且有部分信息被涂抹,更不易读。这正是AI导读可以发挥潜能的场景。
接下来,识林企业会员可在483和警告信页面看到如下图所示的导读:
还需强调的是,AI不可尽信,仅供参考和支持。
另外,会员还可就当前的483和警告信进一步“追问”AI ,具体操作可见用识林AI看483 。
识林® 版权所有,未经许可不得转载
岗位必读建议 QA(质量保证) : 确保所有操作符合DQSA_TITLE_II规定,监控供应链安全。注册专员 : 理解法规要求,确保药品注册过程遵循最新指南。供应链管理 : 遵循产品追溯和验证流程,保障供应链的透明度和安全性。文件适用范围 本文适用于美国境内的化学药品和生物制品,包括创新药和仿制药。涉及的企业包括大型药企、Biotech公司、跨国药企以及CRO和CDMO等。
文件要点总结 供应链安全法案标题 : "Drug Supply Chain Security Act" 明确了药品供应链安全的法律框架。
定义和要求 : 对药品供应链中的术语进行了定义,并规定了制造商、再包装商、批发分销商和配药者的角色和责任。
增强药品分销安全 : 规定了交易信息、交易历史和交易声明的电子交换标准,以及产品标识符的要求。
批发分销商国家标准 : 制定了批发分销商的许可标准,包括存储、处理、记录维护和财务保障等要求。
第三方物流提供商标准 : 对第三方物流提供商的许可和操作标准进行了规定,确保其服务符合联邦法规。
以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位:QA、供应链、注册 QA:立即修订SOP-ABN-QA-0040,补充可疑产品识别、调查流程及24小时报告机制,建立非法产品响应程序。 供应链:更新SOP-ABN-DC-0004,明确盗窃事件上报时限,强化与贸易伙伴的协同调查流程。 注册:评估DSCSA合规差距,协调跨部门整改,确保后续FDA沟通符合时效要求。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :全面核查无菌工艺验证文件,建立干预操作趋势分析系统,修订环境监测程序。 生产 :立即整改无菌操作行为,优化设备清洁与消毒流程,强化人员培训。 工程 :评估并修复灌装线气流与设备损伤问题,验证生物指示剂放置策略。 验证 :重新设计培养基灌装方案,确保其挑战性与商业生产一致。 适用范围 本文适用于无菌及非无菌化学药生产商(如合同生产组织CDMO),涉及FDA监管的美国市场产品,重点关注无菌灌装工艺、环境监测及数据完整性。
总结概要 该483表格记录了FDA对Fareva Amboise无菌生产设施的检查结果,揭示其在无菌保障体系中的系统性缺陷。文件指出无菌工艺验证未充分评估气流模式(如A级区湍流、干预操作导致污染风险),且培养基灌装设计未模拟商业生产的干预频率。环境监测程序存在漏洞,包括未对A级区操作人员实施充分监控、消毒程序缺乏科学验证。设备管理方面,清洁验证未覆盖最难清洁部位,非专用设备存在交叉污染风险。数据完整性缺陷涉及计算机系统无审计追踪、人工修改测试参数。质量管理体系未能确保批记录完整性(如缺失干预记录),且目检程序未建立有效缺陷检测标准。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA (质量保证):需立即审查质量控制部门职责书面化情况,建立未注册实验室的质量协议,强化文件控制体系。 QC (质量控制):核查所有检测数据来源,确保仅使用注册实验室,建立样品转移追溯记录。 生产管理 :核实批生产记录与实际产量的一致性,完善物料平衡追踪。 仓库管理 :制定原料药仓库的接收、存储和分发标准操作规程(SOP),确保环境监测记录完整。 注册 :更新DMF文件,披露所有检测场所,评估合规风险。 适用范围 本文适用于化学原料药(API) 制造商,针对美国市场 的GMP合规要求,涉及印度 监管机构FDA检查发现的问题。适用于大型药企 及跨国制药公司 的原料药生产单元。
文件概要 该FDA 483检查报告记录了印度Hetero Labs Limited第九单元在2025年9月检查中的6项关键缺陷。报告指出质量控制部门未书面化职责,且违规使用未注册实验室检测拟销往美国的原料药,该实验室未在DMF中披露(主要内容1)。检查发现未注册实验室存有与主工厂批号匹配的原料药样品、中间体及未受控检测记录,但工厂无样品转移文件或质量协议(重点内容1)。第二项缺陷涉及原料药及中间体在无接收、存储记录的情况下放行至无证仓库,缺失温度监控和隔离措施(主要内容2)。第三项缺陷揭示批生产记录中原料用量与实际产量不符,仓库中存有未记录的额外批次(重点内容2)。第四项指出质量部门未有效管控GMP文件,包括原始记录被撕毁及签名造假(主要内容3)。第五项缺陷为标签发放与使用未核销,导致批次追溯困难(重点内容3)。最后一项为未调查未注册实验室检测的超标结果,仅记录合格数据(主要内容4)。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA (质量保证):需立即审查质量体系文件,建立书面质量控制程序,强化环境监测趋势分析能力。 生产管理 :整改无菌操作流程,规范人员行为,优化灌装线设计以减少干预。 微生物控制 :重新评估环境监测布点科学性与数据完整性,修订沉降碟使用规范。 验证 :重新设计烟雾试验以匹配商业化生产条件,补充关键干预的气流评估。 IT/数据完整性 :限制LIMS/SCADA系统权限,建立审计追踪审查机制。 适用范围 本文适用于无菌制剂合同生产商 (CDMO),涉及化学药与生物制品的无菌灌装及终端灭菌产品 ,主要针对美国市场 的GMP合规要求。监管机构为美国FDA ,检查聚焦于无菌操作、环境监测、数据完整性及人员培训等核心领域。
文件概要 该FDA 483表格记录了2025年9月对Immacule Lifesciences无菌制剂生产设施的检查结果,共提出8项关键缺陷。文件指出质量体系存在系统性失效,包括质量控制部门未书面定义职责、环境监测趋势分析不充分、无菌操作程序执行偏差(如层流罩消毒不当与物料转移污染风险)。灌装线设计缺陷导致过度人员干预,且烟雾试验未真实模拟商业化生产条件。环境监测布点缺乏科学依据,沉降碟位置被物料遮挡。无菌工艺验证采用“括号法”但未评估所有污染风险,终端灭菌产品的干预未纳入培养基灌装设计。计算机化系统(LIMS/SCADA)权限管控不足,存在数据篡改风险。实验室调查未充分模拟根本原因,部分批次放行未考虑稳定性数据。人员培训体系缺陷体现为目检工具包七年未更新,操作员资质评估未覆盖最差产品条件。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :全面核查设备确认报告与缺陷样品管理程序,确保AQL检测符合21 CFR 211.68要求。 生产 :修订目检规程(SOP P3109),建立缺陷标准库并规范测试套件管理。 注册 :评估未提交FAR的合规风险,更新质量协议。 工程 :重新验证层流设备(PQ/GT2.3.224)并修订干预程序(SOP P366)。 适用范围 本文适用于美国市场无菌注射剂(化学药)生产商,涵盖FDA 483检查发现的设备设计缺陷、无菌控制失效及质量协议缺失问题,涉及跨国CDMO企业。
内容概要 该文件记录FDA对UBI Pharma无菌注射剂生产的检查结果,指出五项关键缺陷。第一,目检设备未针对特定产品缺陷完成确认,缺陷样品库管理不规范,导致AQL检测失效(21 CFR 211.68)。第二,无菌控制程序存在漏洞,包括烟雾研究设计缺陷、操作员干预不当及层流中断处置缺失(21 CFR 211.113)。第三,质量协议过期导致FAR未及时提交,投诉调查不充分(21 CFR 211.198)。第四,变更控制未执行,CAPA有效性不足(21 CFR 211.100)。第五,偏差调查未覆盖已分销批次(21 CFR 211.192)。文件强调企业需系统性整改设备确认、无菌保障及质量体系。
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读指南:
QA(质量保证):确保生产过程符合cGMP规定,保障药品质量与安全。 生产:遵循cGMP标准进行药品生产、加工、包装或储存。 QC(质量控制):进行药品质量检测,确保符合规定标准。 适用范围:
要点总结:
cGMP规定的地位 :强调了cGMP规定是确保药品符合安全、身份、强度、质量和纯度要求的最低标准。合规性与监管行动 :不遵守cGMP规定的药品将被视为掺假,相关责任人将面临监管行动。HCT/Ps的额外要求 :对于HCT/Ps,除了cGMP规定外,还需遵守特定的捐赠者资格和适用的当前良好组织实践程序。适用性与冲突解决 :cGMP规定与其他相关法规相互补充,如有冲突,特定适用于药品的法规优先。临床研究药品的豁免与合规 :I期临床研究药品生产可豁免部分cGMP规定,但进入II期或III期临床研究或合法上市后,必须符合cGMP。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读指南:
QA:负责确保所有操作符合cGMP要求,包括生产、质量控制、设备维护等。 生产:必须遵守书面程序,确保产品质量。 质量控制(QC):负责样品的测试和批准或拒绝,以及稳定性测试。 设备维护:确保设备清洁、维护和校准符合规定。 仓储与分销:遵守药品存储和分发的书面程序。 文件适用范围:
文件要点总结:
质量控制单元的责任: 必须有一个质量控制单元,负责批准或拒绝所有组件、药品容器、包装材料、标签和药品,并审查生产记录以确保没有错误发生或错误已得到全面调查。
人员资质与责任: 参与药品生产、加工、包装或储存的人员必须具备相应的教育、培训和经验,并遵守良好的卫生习惯。
设备设计、清洁与维护: 设备应适当设计,便于操作、清洁和维护,并按规定进行定期清洁和维护。
组件和药品容器的控制: 必须有书面程序详细描述组件、药品容器和闭合件的接收、识别、存储、取样、测试和批准或拒绝。
生产和过程控制: 必须有书面程序确保药品具有其声称或代表的身份、强度、质量和纯度,包括偏差处理和产量计算。
包装与标签控制: 必须有书面程序确保正确的标签和包装材料用于药品,包括防篡改包装要求。
仓储与分销程序: 必须有书面程序描述药品的存储和分发,确保药品质量。
实验室控制: 必须建立科学合理的规格、标准、抽样计划和测试程序,以确保药品及其组件符合适当的身份、强度、质量和纯度标准。
记录与报告: 所有与生产、控制或分发相关的记录必须保存至少一年,或在特定情况下保存更长时间,并随时可供授权检查。
退回和报废药品的处理: 退回的药品必须被识别并保留,除非证明其符合适当的安全、身份、强度、质量和纯度标准,否则应销毁。
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
研发(R&D) :应深入理解IND的提交要求、临床试验阶段及相应变更流程,确保研发过程中符合FDA规定。临床(Clinical) :必须熟悉临床试验的各个阶段,包括患者入组、数据收集和安全报告等,以保障试验顺利进行。注册(Regulatory Affairs) :需掌握IND的提交流程、年度报告和安全报告的要求,以确保合规提交。质量管理(QA) :应监督整个IND流程,确保所有记录和报告符合FDA规定,并在必要时进行审计。文件适用范围:
本文适用于在美国进行的临床研究,涉及化学药、生物制品等药品类型,包括创新药、仿制药、生物类似药等注册分类。适用于Biotech、大型药企、跨国药企等企业类别。由美国食品药品监督管理局(FDA)发布。
文件要点总结:
IND申请要求 :明确了提交IND的必要条件,包括药物的临床试验阶段和IND内容格式。临床试验阶段 :将临床试验分为三个阶段,每个阶段都有其特定目的和要求。IND内容和格式 :详细列出了IND应包含的内容,如药物信息、临床试验方案、药物的化学制造与控制信息等。安全报告 :强调了对不良事件的监测和报告流程,确保患者安全。IND的变更和年报 :规定了对IND进行变更的流程和年度报告的要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :全面审核偏差调查程序,强化质量体系监督,确保CAPA有效性。 生产 :严格执行无菌操作SOP,升级设备维护计划,落实环境监测整改。 工程 :评估设备生命周期管理,制定预防性维护方案,消除污染风险。 注册 :评估违规对申报文件的影响,确保整改措施符合FDA要求。 适用范围 本文适用于在美国市场销售的无菌化学药(如眼科溶液、喷雾剂)的生产企业,涉及CGMP违规问题(21 CFR 210/211),重点针对无菌工艺、偏差调查及设备维护缺陷。
总结 该警告信指出Apotex公司无菌药品生产存在三项重大CGMP违规。第一,偏差调查系统失效,包括未及时调查泄漏测试失败、容器密封性缺陷及稳定性样品问题,且未评估对已分销批次的影响。第二,无菌控制不足,表现为隔离器清洁维护缺陷、手套破损应急处理不当、环境监测设计不合理。第三,设备管理失当,如颗粒物污染未及时解决,升级延迟。FDA要求企业提供独立评估报告及整改计划,涵盖偏差调查系统、CAPA程序、变更管理及无菌操作风险分析。企业已召回部分产品并暂停生产,但需系统性改进质量体系,包括加强质量部门权限及管理层监督。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :强化偏差调查程序,确保根因分析和CAPA有效性;建立缺陷类型独立监控机制。 生产 :优化无菌操作流程,修订设备清洁验证协议;实施供应商来料增强检测。 供应链 :修订供应商质量协议,增加关键组件(如胶塞)的抽样标准和频次。 注册 :评估违规对上市许可的影响,准备与FDA的合规沟通材料。 适用范围 本文适用于在美国市场生产无菌制剂(如注射剂)的跨国药企及CDMO,涉及化学药与生物制品的商业化生产。监管机构为FDA,违规聚焦CGMP(21 CFR 210/211),问题包括胶塞污染、无菌工艺缺陷及偏差调查不足。
总结 该警告信指出Catalent Indiana工厂在CGMP合规性上的系统性缺陷,主要涉及偏差调查不足和无菌控制失效。具体问题包括对胶塞携带哺乳动物毛发污染的调查不充分,未及时追溯污染源至供应商,且未有效实施CAPA;无菌工艺中未验证关键设备(如胶塞安装部件)的清洁效果,环境监测方法(接触皿替代拭子)不足以检测微生物污染。FDA要求企业提交独立评估报告,涵盖偏差调查体系全面整改、供应商质量协议修订、无菌工艺再验证,以及污染风险的全设施评估。文件强调需回溯审查客户投诉数据,并制定数据驱动的持续改进计划。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :强化供应商审计程序,建立供应商动态监控机制,确保稳定性数据合规性。 供应链 :完善供应商准入与退出标准,验证运输条件对API质量的影响。 生产 :严格执行放行前测试流程,禁止未经QU批准的物料流转。 注册 :核查NDC清单与SPL一致性,确保API注册信息准确。 适用范围 本文适用于美国FDA监管的化学原料药(API)生产企业,涉及供应商管理、稳定性数据、标签合规性及CGMP违规问题,重点关注跨国药企及Biotech的供应链合规风险。
总结 该警告信指出Darmerica, LLC在API生产中的多项CGMP违规行为,包括质量体系失效、供应商管理缺陷及标签错误。文件首先概述了企业因质量单元(QU)未履行监管职责导致API掺假的违法事实,具体表现为供应商评估不足(如接受虚假检查声明)、未调查质量投诉(如未追溯污染批次的其他客户)及放行前测试缺失(如提前分销未完成分析的API)。其次,文件详述了标签误导问题(如隐瞒GLP-1 API真实名称)和注册违规(如未正确提交510(j)清单)。FDA要求企业在15个工作日内提交系统性整改计划,涵盖供应商重审、客户通知、QU职能强化及第三方审计。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QA :全面审核偏差调查流程,强化CAPA有效性验证,建立微生物监控升级机制。 生产 :优化洁净区维护计划,修订设备清洁SOP,加强人员无菌操作培训。 QC :复核历史OOS结果无效化决策的科学依据,完善实验室调查程序。 工程 :评估HVAC系统与制药用水系统的设计缺陷,制定紧急修复方案。 适用范围 本文适用于美国市场无菌化学注射剂生产企业(含CDMO),涉及终端灭菌工艺缺陷、微生物/内毒素控制失效及设施设备合规问题。监管机构为FDA。
总结概要 该警告信基于2025年FDA对Liebel-Flarsheim公司的现场检查,指出其违反21 CFR 210/211的CGMP要求,具体涉及两大核心问题。第一,企业未彻底调查40余起生物负载超标(含TNTC结果)及内毒素OOS事件,调查结论缺乏科学依据,CAPA措施延迟至2026年完成,且过度依赖终端灭菌工艺而忽视过程控制风险。第二,设施设备存在系统性缺陷,包括洁净区维护不足(HEPA密封胶带修补、设备锈蚀)、制药用水系统腐蚀泄漏,且环境监测长期显示微生物污染,但整改计划延至2027年。FDA要求企业提交独立回顾性评估报告,涵盖所有无效化OOS结果的实验室与生产根本原因分析,并全面整改微生物控制体系、设施设计及水系统。监管措施包括可能扣留出口证书、暂停新申请审批及启动产品召回。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位 必读岗位 :临床研究者(PI)、临床监查员(CRA)、临床研究协调员(CRC)、医学写作(MW)。工作建议 :
PI :需严格审核受试者入组标准,确保与方案一致,建立双重核查机制。 CRA :加强现场监查,重点核查入组文件与源数据一致性。 CRC :更新知情同意流程,确保与修订后方案匹配。 MW :跟踪方案变更,及时更新文件并同步IRB。 适用范围 本文适用于美国FDA监管的化学药或生物制品临床试验,涉及创新药或改良型新药,主要针对临床研究机构(如医院、学术中心)及申办方(Biotech或大型药企)。
文件概要 该警告信指出临床研究者在执行两项糖尿病药物临床试验时违反21 CFR 312法规,具体涉及未遵循研究方案和未及时向IRB报告变更。第一项违规为入组7名不符合血压、eGFR或禁用药物(SGLT2抑制剂/GLP-1受体激动剂)标准的受试者,研究者虽事后修改方案并获IRB批准,但滞后超过一年,且纠正措施未充分说明未来如何确保监督。第二项违规为未及时报告研究设计从双盲随机试验变更为包含预试验的两阶段研究,导致所有受试者均接受试验药物且知情同意文件未同步更新。FDA强调研究者需确保方案依从性以保护受试者权益和数据完整性,并要求15个工作日内提交详细整改计划。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议 必读岗位 :
QC :强化OOS调查程序,建立微生物污染溯源机制,确保CAPA有效性评估。 QA :完善质量体系文件,明确QU职责范围,加强实验室操作及偏差管理的监督。 实验室管理层 :审核历史OOS数据,修订调查流程,落实独立回顾性审查。 适用范围 本文适用于为美国市场提供化学药及OTC产品检测服务的合同检测实验室(CDMO),涉及CGMP合规性缺陷,主要针对微生物检测与质量体系漏洞。
总结 该警告信指出受检实验室存在两项关键CGMP违规:未彻底调查微生物检测OOS结果(21 CFR 211.192),以及质量部门未履行书面程序及监督职责(21 CFR 211.22(d))。具体问题包括多次以“未知实验室错误”草率结案,未科学论证污染源即依赖复测数据,且CAPA未评估有效性;质量体系缺乏偏差管理、变更控制等核心程序。FDA要求提供三年内OOS结果的独立回顾性分析报告,并提交质量体系全面评估及整改计划,涵盖QU职能强化、实验室趋势监控及调查流程修订。文件援引FDA质量体系指南,强调需通过风险管理实现CGMP合规。
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。