首页
>
资讯
>
AI 参与临床试验、申报资料和偏差调查,要告诉药监局吗?
出自识林
AI 参与临床试验、申报资料和偏差调查,要告诉药监局吗?
2025-03-24
年初DeepSeek流量爆发以来,各行各业“接入”蔚然成风,医药行业也不例外。据报道,短期内就有上百家医疗机构接入DeepSeek,且其中多为地方龙头医院。药企方面,最早是网上流出恒瑞红头文件要求全公司范围内开展DeepSeek应用工作,随后信达、君实等药企也纷纷公布自家AI项目(本文AI均指基于大语言模型的AI)。这其中也有识林AI项目团队的参与 。
医疗和医药均是众所周知的高度监管行业,AI热潮的背后,不可避免的一个话题就是监管。近日,一项发表在JAMA Network Open上的研究《患者对于AI起草的电子信息回复偏好的伦理考量》(Ethics in Patient Preferences for AI-Drafted Responses to Electronic Messages)揭示了AI在医院应用中的挑战,联想到我国刚刚发布的AI标识管理办法 ,以及FDA在1月份发布的首个制药AI指南 ,药企可考虑为即将到来的AI监管做好准备。
一旦得知AI参与,患者满意度下降,与其质量无关
近年来,患者通过电子门户向医生发送消息的数量不断增加,这给医生带来了巨大的工作负担。为缓解这一问题,美国许多医院就尝试使用AI技术来生成消息回复。
研究团队在杜克大学卫生系统的患者咨询委员会 中开展了一项调查研究,共有2511名成员参与,其中1455人完成了调查。参与者被随机分配到不同的实验场景中,分别评估了消息的严重性(如常规药物续订请求、药物副作用问题以及影像学检查发现恶性肿瘤的可能性)、作者身份(AI或人类医生)以及是否披露来源信息(AI、人类医生或不披露)对患者满意度的影响。
研究结果显示:
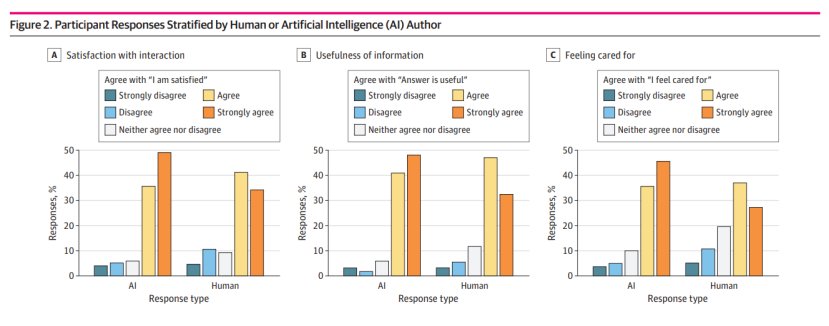
AI撰写的回复更受青睐:研究发现,患者对AI撰写的回复的整体满意度、信息有用性以及感受到的关怀程度均高于人类撰写的回复。具体而言,AI撰写的回复在满意度上平均高出0.30分(满分5分),在信息有用性上高出0.28分,在感受到的关怀程度上高出0.43分。
消息严重性不影响偏好:研究还发现,无论消息的严重性如何(从常规药物续订到潜在的恶性肿瘤),患者对AI撰写的回复的偏好并未因消息的严重性而改变。
披露AI使用会降低满意度:尽管患者对AI撰写的回复本身较为满意,但当明确告知他们回复是由AI生成时,满意度会略有下降。研究显示,与披露是人类医生作者相比,披露AI的满意度平均下降了0.13分;与不披露相比,披露AI的满意度平均下降了0.09分。(见下图)
研究结果揭示了一个伦理困境:尽管患者对AI撰写的回复本身较为满意,但患者可能更倾向于认为消息是由人类医生撰写的,且对AI的介入感到不安。然而,从伦理角度来看,患者有权知道他们所接收的信息是由AI生成的,这关系到患者的自主权和知情权。
研究者指出,尽管披露AI使用可能会导致患者满意度略有下降,但这不应成为阻碍披露的理由。研究中,患者最倾向于接受的披露方式是:“此消息由医生在自动化工具的支持下撰写。”这种简短的披露方式强调了人类专家的主导,既能尊重患者的知情权,又不会因过多的技术细节而让他们感到困惑。
披露只是基本要求,重要的是AI该如何取信于药监
从上述医疗AI应用的研究中,制药行业需要考虑的第一个问题是披露。但可想而知,披露是未来AI合规的基本要求。3月15日,国家互联网信息办公室发布《人工智能生成合成内容标识办法》 ,旨在规范人工智能生成合成内容的标识,自2025年9月1日起施行。所以药企首先面临的问题在于,到底哪些流程和文件需要向监管部门披露AI的参与,哪些又不需要?
第二个启示在于,之所以出现上述研究结论,其本质是人类目前还无法完全信任AI,这不仅仅是出于某种“歧视”,也是基于日常使用AI处理专业工作时发现的种种问题,典型的例如“AI幻觉”。那么AI该如何取信于人类?从药品监管角度看,AI不但要取信于药企,还要取信于药监。
迄今为止最具参考价值的答案在1月份FDA 发布的《使用人工智能支持药品和生物制品监管决策的考量》 指南草案中。作为全球主要监管机构对AI监管的最新考量,涵盖广泛的AI技术,并且为企业提出了具体可执行的AI应用评估框架。
该指南草案中,提出可信度(credibility)概念,定义是通过收集可信度证据而建立的对AI模型在特定使用场景(context of Use, COU)中的信任。COU则明确了AI模型用于解决特定问题时的具体角色和范围。AI模型的可信度评估应与模型风险相称,并针对特定的COU进行调整,以确保AI模型输出在监管决策中的适用且可靠。
指南草案中,FDA提出了一个“七步走”的可信度评估框架,指导企业评估AI模型的可信度,其中实操的关键在于第3和第4步。
第1步:定义感兴趣的问题(question of interest)
第2步:定义AI模型的使用场景
第3步:评估AI模型风险
此处模型风险由模型影响和决策后果两个因素决定。模型影响指AI模型提供的证据相对于其他证据的贡献程度,即是否单独用于某项决策,且无其他信息配合;决策后果描述因错误决策导致的不利结果的重要性,这个因素完全与AI技术独立开来,仅考虑对药品安全、有效和质量的影响。这个双因素模型简明扼要,避开对复杂AI技术的探讨,将评估重点聚焦于决策的过程和影响。
第4步:制定建立AI模型可信度的计划
FDA要求企业对AI模型开展技术层面的评估,其维度与其他监管机构类似,重点在数据和训练。尽管基于大语言模型的AI是众所周知的“黑箱”,FDA还是针对模型测试提出了相当具体的要求。
该计划需详细描述模型及其开发过程,包括模型的输入、输出、架构、特征及其选择过程、模型参数等,并解释选择特定建模方法的理由。同时,要详细说明用于开发模型的数据集,涵盖数据的收集、处理、标注、存储、控制和使用情况,确保数据相关且可靠,即包含关键数据元素、具备足够代表性,且准确、完整、可追溯 。此外,还需描述模型的训练过程,包括学习方法、评估模型性能的指标、防止过拟合或欠拟合的技术、训练超参数等,并说明是否使用了预训练模型以及模型的校准情况。
在模型评估方面,要说明测试数据的收集、处理、标注、存储、控制和使用情况,确保测试数据独立于开发数据,以准确评估AI模型的性能。同时,需描述测试数据对使用场景的适用性,考虑数据漂移现象,即开发数据与部署环境中遇到的数据可能不同,导致模型性能变化。还要描述模型预测与观测数据之间的一致性,解释选择的模型评估方法的理由,提供用于评估模型的性能指标,并包括置信区间 。此外,要说明模型预测不确定性和置信水平的估计过程,以及任何其他量化置信度或不确定性的描述或指标,描述建模方法的局限性,包括潜在的偏差 ,并详细说明代码验证的质量保证 和控制程序。
第5步:执行计划
第6步:记录可信度评估计划的结果并讨论偏差
第7步:确定AI模型对使用场景的适用性
此外值得一提的是,该指南以对药品的安全、有效和质量的影响为标准,明确不涵盖以下两类AI应用:药物发现阶段的AI应用;用于提高操作效率(如内部工作流程、资源分配、起草/撰写申报资料)的AI应用。可见FDA认为这些应用场景对患者安全、药品质量影响风险较低。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位:
RA(注册) :必读,需理解AI在药品和生物制品注册决策中的应用和要求。R&D(研发) :必读,应掌握AI技术在药品和生物制品研发中的应用指导。QA(质量管理) :必读,了解AI技术如何影响质量管理和合规性。工作建议:
RA:评估现有注册流程中AI技术的适用性,确保符合FDA的最新指导。 R&D:在研发过程中考虑AI技术的整合,提高效率和准确性。 QA:监控AI技术在质量管理中的应用,确保符合监管要求。 适用范围:
文件要点:
AI技术应用: 明确了AI技术在药品和生物制品监管决策中的潜在应用,强调了其在提高决策效率和准确性方面的重要性。数据治理: 强调了在使用AI技术时,必须确保数据的质量和治理,以支持有效的监管决策。透明度和可解释性: 特别强调了AI模型的透明度和可解释性,要求企业能够解释AI决策过程。风险管理: 提出了对AI技术相关风险的管理要求,包括对算法偏差和错误结果的识别和控制。监管互动: 鼓励企业与FDA进行沟通,以确保AI技术的应用符合监管要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
市场/医学传播 (必读):审核AI生成的宣传材料、患者教育内容是否添加显式标识,确保元数据合规。 IT/数字化团队 (必读):与外部AI服务商合作时,要求其提供标识合规证明;内部AI工具需嵌入标识功能。 合规/法务 :更新用户协议,明确AI生成内容标识义务;留存相关操作日志。 以上仅为部分要点,请阅读原文,深入理解监管要求。