首页
>
资讯
>
EDQM 站在药企角度梳理 CEP 申报流程
出自识林
EDQM 站在药企角度梳理 CEP 申报流程
2025-11-11
欧洲药品质量管理局(EDQM)于11月5日发布了一份指南文件《获得CEP/变更批准的分步流程》 ,梳理了申请欧洲药典适用性证书(CEP )以及变更 审批的分步流程。
该文件简明扼要但却非常贴心。据负责CEP申请的识林向导分享,文件中的内容对于行业老手来说比较熟悉,但对于刚接触CEP申报的新人则有独特价值。如今各类法规指南文件越来越多(尤其是存在多头监管的欧盟),有时令药企有些无所适从,EDQM跳出监管思维,从药企办事流程开始梳理,值得点赞。
在文件中,EDQM将新申请(New Applications)、姊妹文件申请(Sister Files,同分子CEP快速路径)、修订(Revision)、通知(Notification)、续期(Renewal)以及变更持有人 (Transfers of holdership)等不同类型的CEP申请统一对应到同一套标准化流程中。即无论何种申请,对应的都是:申报(Submission)、验证 (Validation)、审评或处理(Assessment or treatment)以及结果沟通(Communication of the decision)这四个阶段,流程也基本相同,区别在于每个阶段的要求不同,对应的时限也有区别。
EDQM重点强调验证阶段,包括格式验证和内容验证。前者主要检查电子CEP申请提交是否符合要求;后者则确保申请完整,足以进入下一步审评流程,如有必要,EDQM会要求补充数据。验证成功后,EDQM会发送收到确认函,注明评估时间表。新申请在通过初步验证后,无论后续结果如何,均需支付费用。从描述上看,这一步类似于我国的“注册受理审查”和“立卷审查”的结合。
接着强调的是审评完成后的结果沟通阶段。必要时,EDQM将根据问题严重程度,分别发出补充信息请求(“发补”)、澄清请求或文件更新请求。为确保高效,单个CEP流程最多有两次信息请求。
文件还特别强调了CEP程序中评估与GMP 检查活动之间的互动。对于相关场地,检查可能在CEP 授予前或后进行。若新申请或修订涉及已被判定GMP不合规的场地,程序将暂停,等待批准前检查 确认其符合欧盟GMP要求。
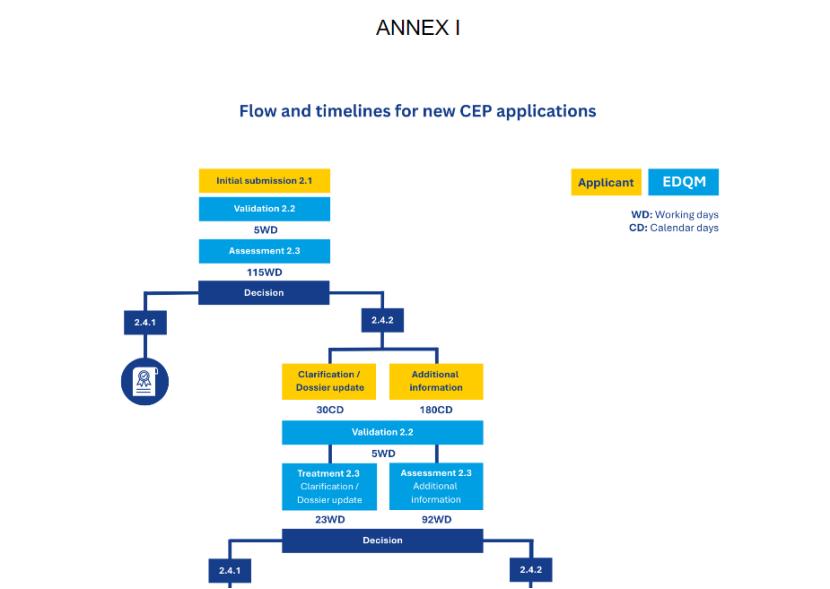
EDQM在附件中提供了多个流程图,详细列出了不同申请类型各阶段的时间表,以工作日(WD,不含周末、公共假期和EDQM假日)或日历日(CD)直观表示,如题图所示。具体可见文件附录。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
QA(质量保证):必读。需确保提交的CEP申请符合EDQM要求,并监控整个申请流程的合规性。 注册(Regulatory Affairs):必读。负责提交CEP申请,并与EDQM沟通,确保申请流程顺利进行。 R&D(研发):必读。需确保提供的数据包符合要求,并在需要时提供补充数据。 GMP(生产质量管理规范):必读。与EDQM检查活动互动,确保生产/分销场所符合GMP和CEP申请要求。 文件适用范围:
文件概要:
以上仅为部分要点,请阅读原文,深入理解监管要求。