|
首页
>
资讯
>
EFPIA 发 GMP 检查年报,全球互认仍待改进
出自识林
EFPIA 发 GMP 检查年报,全球互认仍待改进
2026-05-25
近日,欧洲制药工业协会联合会(EFPIA)发布报告《2025年度监管 GMP/GDP 检查调查数据》。该调查自2003年启动,核心目的并非分析检查缺陷,而是通过统计数据推动检查依赖(inspection reliance)的落地,使得PIC/S成员资格和双边互认协议(MRA)为药企减轻更多负担。
报告数据来自艾伯维、阿斯利康、百时美施贵宝、礼来、默沙东等全球主要跨国药企的反馈。EFPIA提醒读者注意报告数据的局限性。
EFPIA的发现和呼吁:PIC/S成员应更多互认,药企应更主动申请
EFPIA在报告开头罗列3个核心发现:
- 境外检查持续下降:监管机构正越来越多地依赖彼此的检查结果,重复检查正在减少。
- 检查更聚焦、要求更高:重点转向系统化、基于风险的评估和针对性提问,而非简单的程序检查。
- 操作层面挑战犹存:各国检查标准解读和报告格式差异仍待协调;突击检查主要影响组织协调(logistics)。
具体来看,关于药品生产场地检查(见报告第7页):
- 未有任何场地报告因检查导致供应中断。“有因检查”数量在减少,且同样未造成供应中断。预先通知检查与突击检查的结果相似。
- 突击检查主要由中国和美国FDA执行。多个检查机构(如日本、欧盟、美国、韩国)执行批准前检查(PAI)。
- 境外检查:报告检查数量下降,显示出对依赖检查机制的积极信号。23年来首次没有新的国家被报告执行境外检查(即没有新的国家监管部门开展境外检查)。
报告向监管机构和行业提出三项核心行动建议:
1. 监管机构应系统化使用PIC/S等依赖机制,推动已建立贸易关系的监管机构之间扩大互认范围。
2. 加强协调:在检查排期、范围对齐、统一报告格式等方面推进合作,扩大对可信监管机构(尤其是PIC/S成员)的认可。
3. 药企应主动识别潜在的重复检查情形,并正式向监管机构申请依赖。
部分数据和结论概要
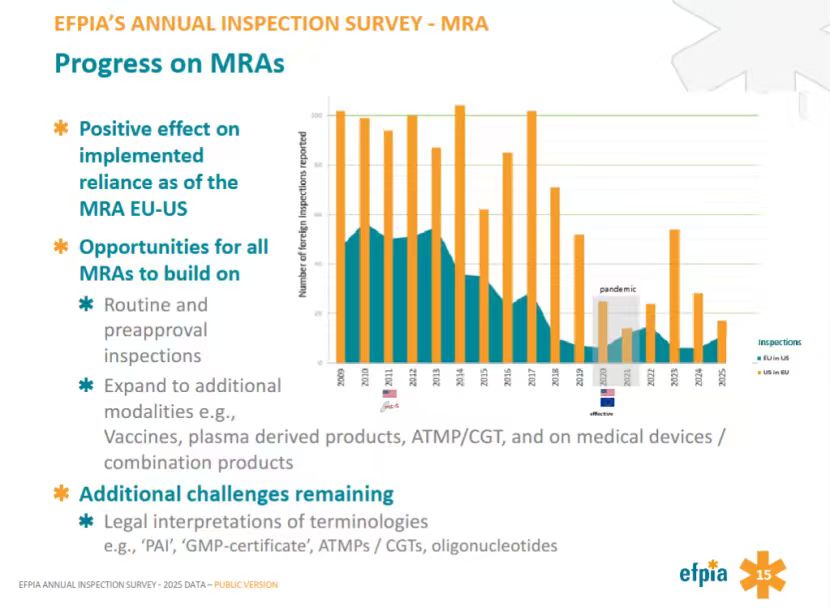
欧美互认成效显著,下一步是互认PAI
从第15页数据的发展趋势来看,在2009年至2019年的MRA实施前期,双边跨国检查较为频繁,尤其是美国FDA在欧盟境内的检查长期维持在每年80至100次以上的高位(即使FDA于2011年加入PIC/S)。随着美欧双方逐步推进GMP检查互认,双边的重复检查数量开始出现显著且持续的下降,其中因全球新冠疫情和旅行受限导致检查数量跌至历史低谷,且在2023年出现过一次短暂的反弹,但整体在2024和2025年再次呈现下降趋势。
但EFPIA认为MRA仍有巨大的优化和拓展空间,未来可以推动将互认范围从日常例行检查进一步扩大到PAI,并争取将互认范围延伸至更复杂的生物制品和先进疗法。另一主要的挑战在于各方对法规术语的法律合规性释义存在差异,例如“PAI”、“GMP证书”以及“寡核苷酸”等概念仍需进一步达成共识。
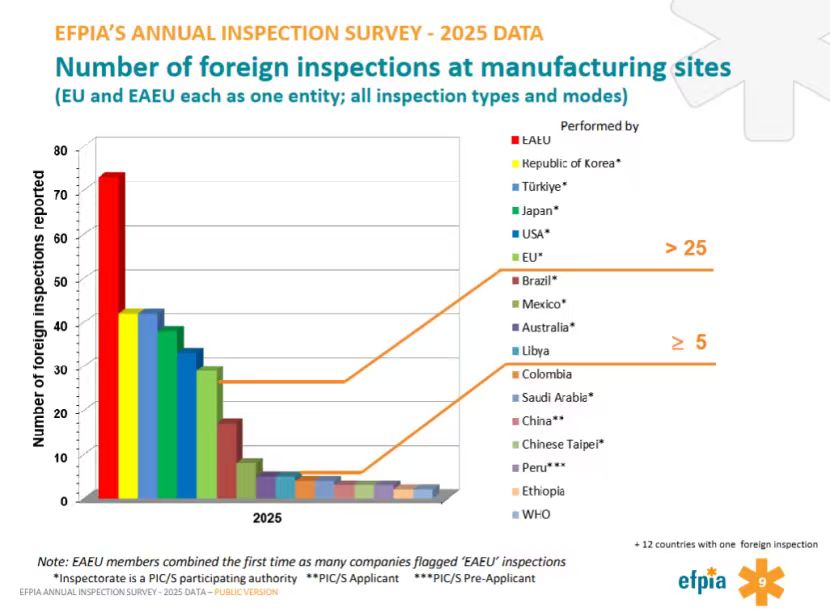
欧亚经济联盟、韩国和土耳其海外检查多于美欧
根据第9页的数据,欧亚经济联盟(EAEU,含俄罗斯,白俄罗斯和中亚多国)首次作为整体被纳入统计,反映出该区域监管协作正在加强。执行海外检查数量名列前茅的监管机构还有韩国、土耳其、日本,超过美欧。除了排名第一的EAEU,多数检查执行方为PIC/S正式成员,中国为申请者,秘鲁为预申请者。
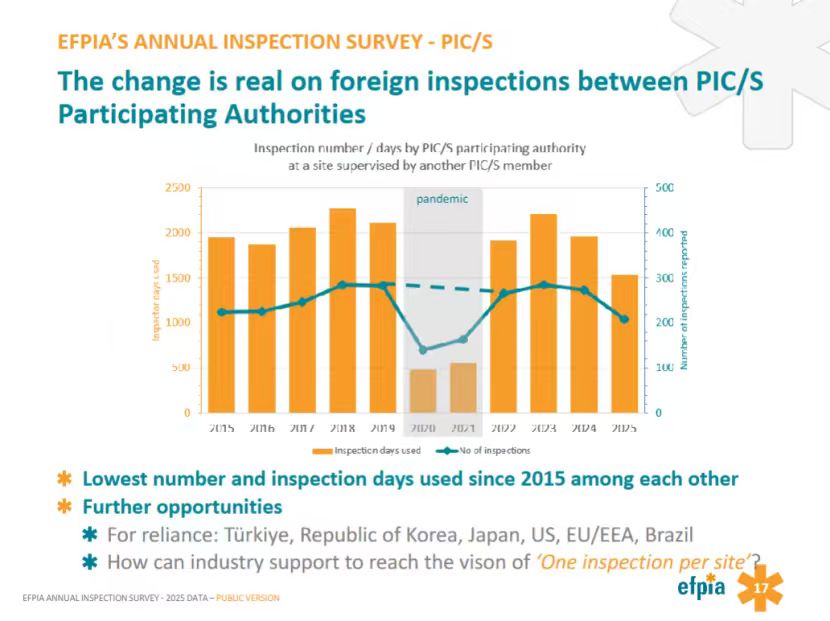
PIC/S成员之间互认仍有较大空间
从第17页的数据来看,在2015年至2019年期间,PIC/S成员机构之间的跨国重复检查整体维持在高位,检查次数稳步攀升至每年近400次,年均消耗的检查天数也突破了2000天。在2020年至2021年期间,受全球新冠疫情和跨境旅行限制的严重影响,现场检查活动急剧下跌,检查天数骤降至500天左右。随着疫情结束,检查活动在2022年和2023年出现了明显的反弹,检查次数和天数再度逼近疫情前的峰值。这一反弹趋势在2024和2025年得到了有效扭转,检查次数和所消耗的天数均表现出一定幅度的回落,2025年的数据更是达到了除疫情特殊时期之外,自2015年以来的历史最低水平。这一变化表明,PIC/S成员机构之间的监管互认举措正在切实发生并取得成效。
EFPIA指出未来在监管互认方面仍有进一步加强互信的空间,特别是在土耳其、韩国、日本、美国、欧盟/欧洲经济区和巴西等国家与地区之间。目前距离实现“一个场地,一次检查”(One inspection per site)的愿景还任重道远。
监管之间存在沟通问题,药厂遭冗余检查
第21页是总结性“行动呼吁(Call to Action)”,指出目前重大障碍,重点阐述如何进一步促进和落实药品检查的监管互认。
EFPIA提醒药企应当更加主动地申请监管互认。这要求企业积极识别可适用互认的场景,并制定企业内部指南来规范和推进此类互认申请。一个典型的例子是:某工厂在一年内先后接受了PIC/S成员机构新加坡卫生科学局(HSA)和欧盟成员国监管机构的检查。
EFPIA认为目前阻碍互认全面落地的三大障碍:
- 许多监管机构仍倾向于用传统、保守的方式去解读现行的法律法规要求,缺乏灵活性;
- 在评估检查报告时存在明显的术语障碍,例如报告中未明确提及“PAI”字样,或者对检查范围的文字表述存在细微差异,都容易导致互认受阻;
- 监管机构内部的硬性指导原则往往形成限制,例如某些机构的内部指南强制规定本机构必须至少每5年对生产场地进行一次现场检查。
药企更看重监管合作和联合检查
第20页显示,被问及“监管部门的哪类合作带来最大实效?”,EFPIA的会员企业认为最有利的是监管机构间加强协调,扩大监管互认框架的应用范围;其次是更多经协调的或联合检查(如ICMRA的协作混合检查试点),统一检查范围并共享报告;排在最后的是MRA双边协议,以及PIC/S、ICH成员间的协作。
识林-实木
识林®版权所有,未经许可不得转载
法规指南解读 适用岗位: 工作建议: - QA:确保所有生产活动符合GMP要求,监督无菌药品生产流程。
- 生产:按照GMP要求执行无菌生产操作,确保产品质量。
- 研发:在药品开发阶段考虑GMP合规性,设计符合要求的生产流程。
- 临床:确保临床试验用药的无菌性和质量符合GMP标准。
- 注册:在药品注册过程中提供符合GMP要求的生产和质量控制信息。
适用范围:
本文适用于化学药品、生物制品的无菌药品生产,包括原料药、制剂等。适用于欧盟地区的Biotech、大型药企、跨国药企等。 要点总结: - 无菌药品生产环境:强调了对无菌药品生产环境的严格控制,包括洁净室的分类和设计,以及对生产环境的持续监测。
- 质量风险管理(QRM):在整个文件中,QRM是确保无菌药品生产质量的核心原则,要求企业在设计和控制生产设施、设备、系统和程序时应用。
- 关键控制点:提出了无菌药品生产过程中的关键控制点,包括设施设计、设备操作、过程验证、环境监测和人员培训等。
- 污染控制策略(CCS):强调了CCS在无菌药品生产中的重要性,要求企业实施全面的CCS以确保产品质量和安全。
- 无菌工艺验证:要求对无菌工艺进行验证,包括无菌过程模拟(APS)和其他相关测试,以确保生产过程能够持续产生无菌产品。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |