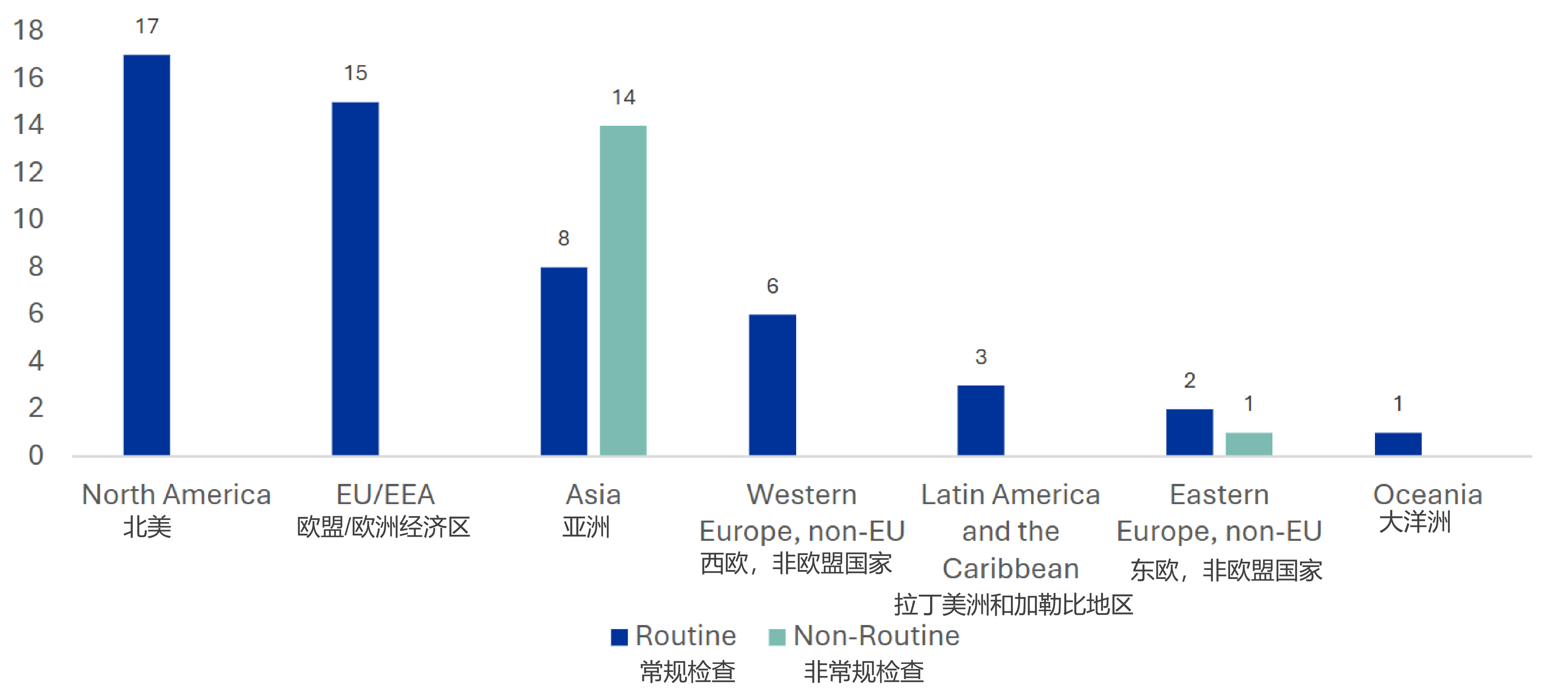
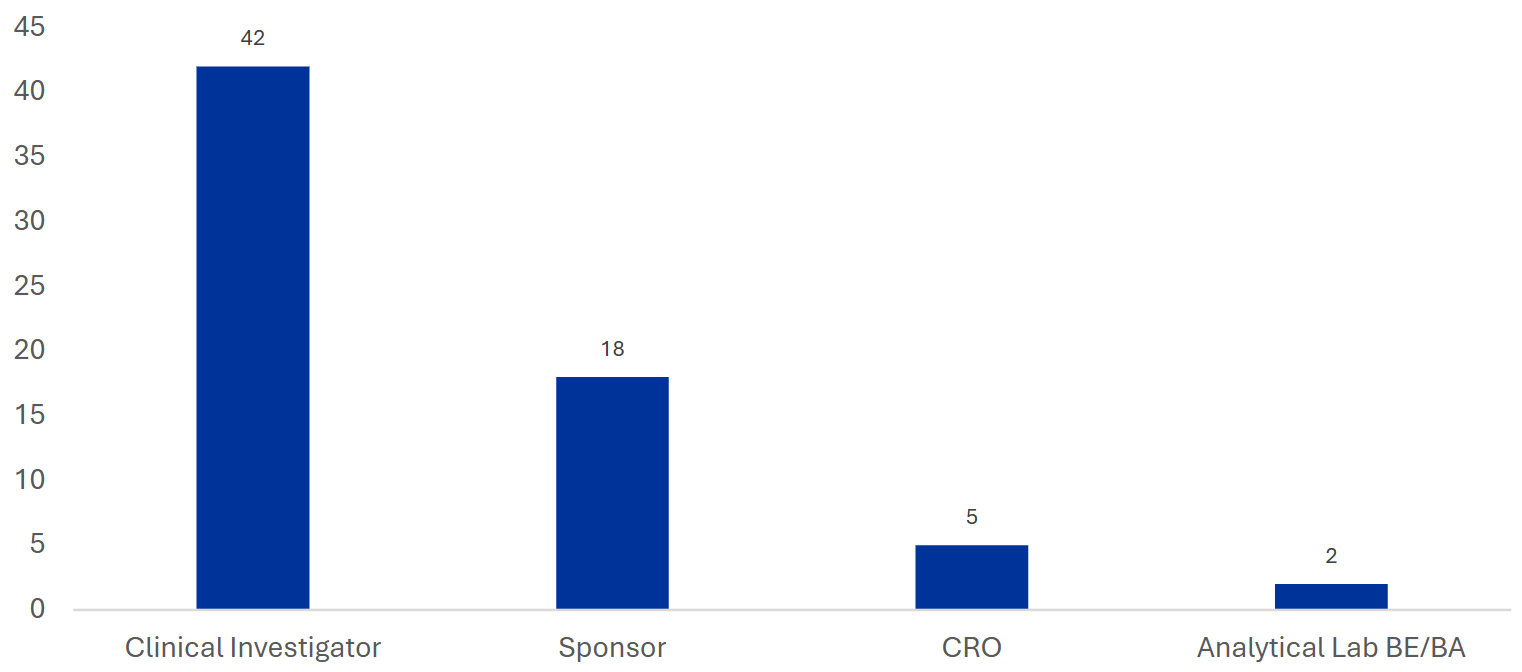
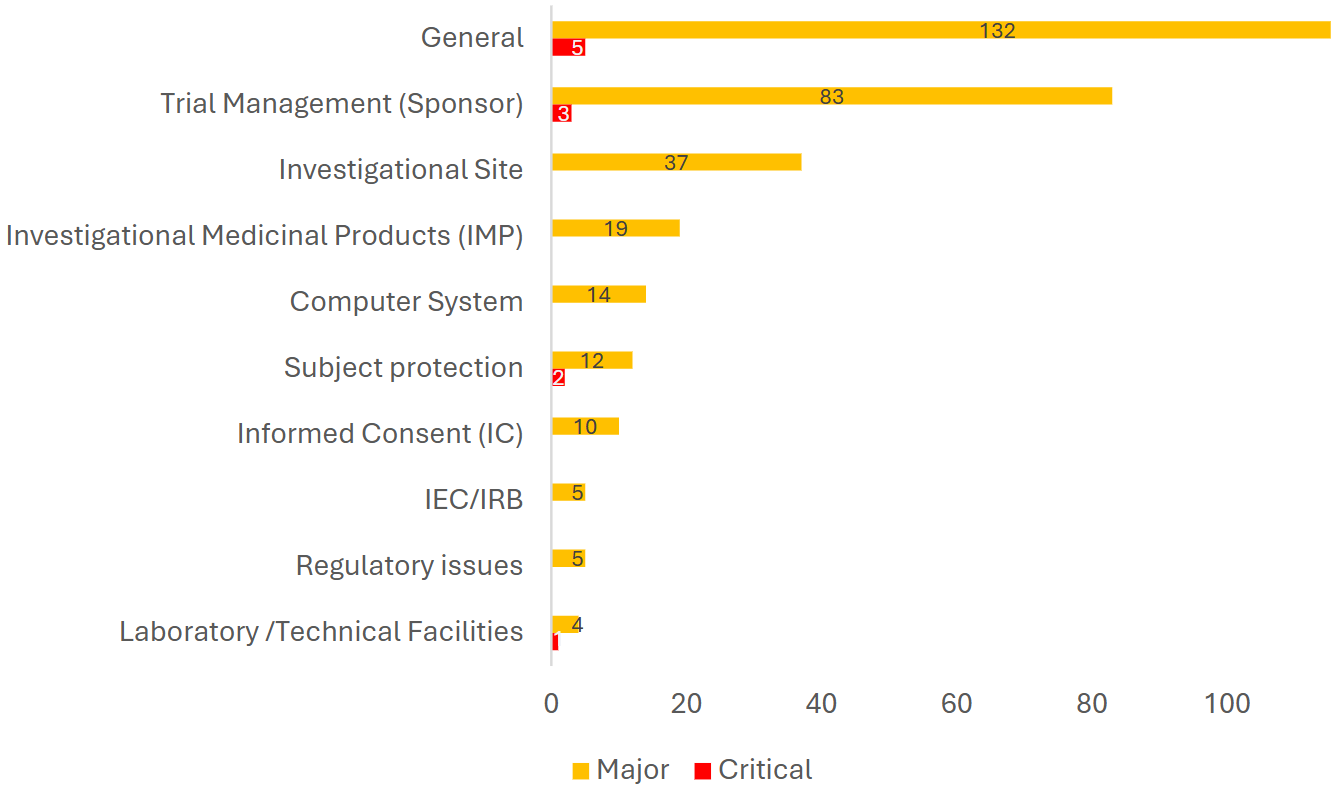
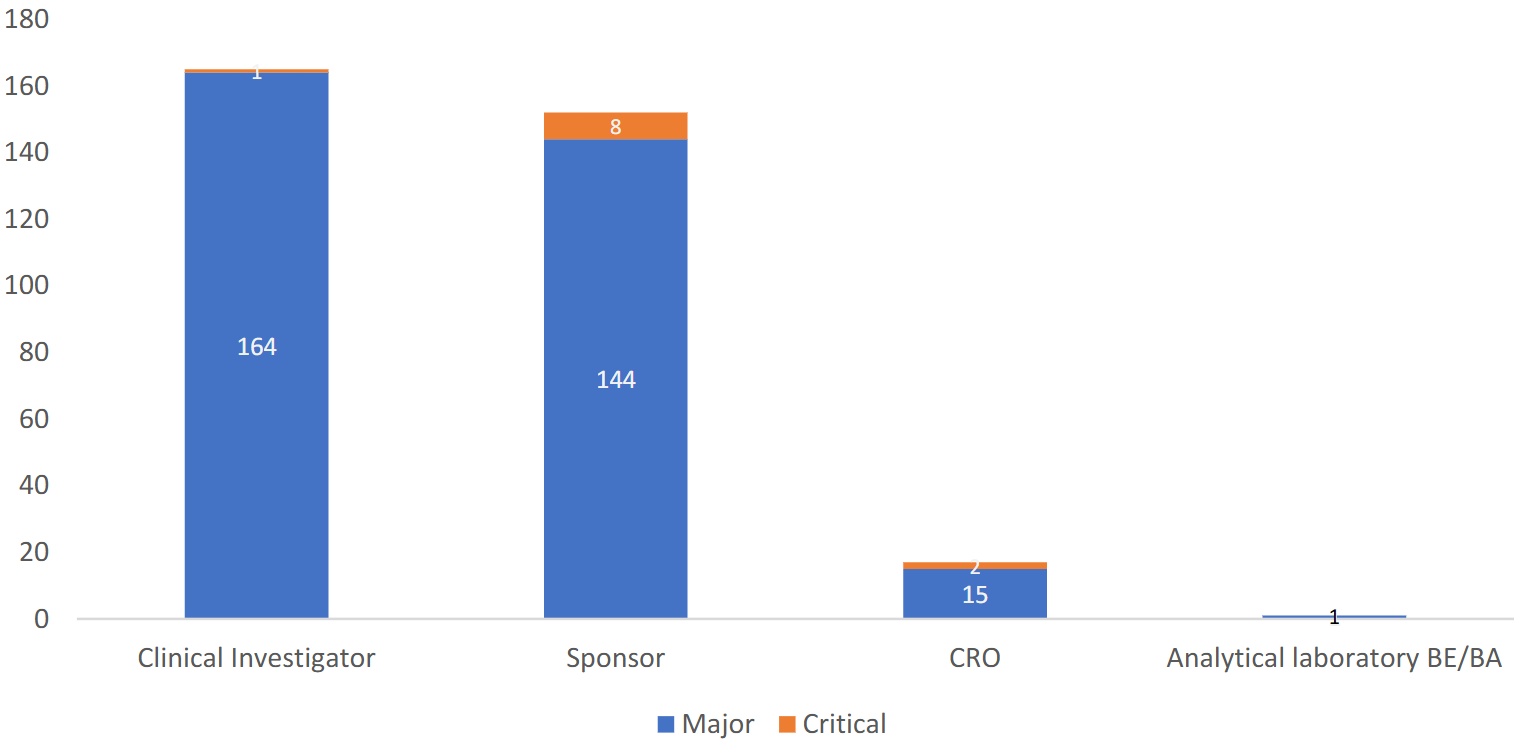
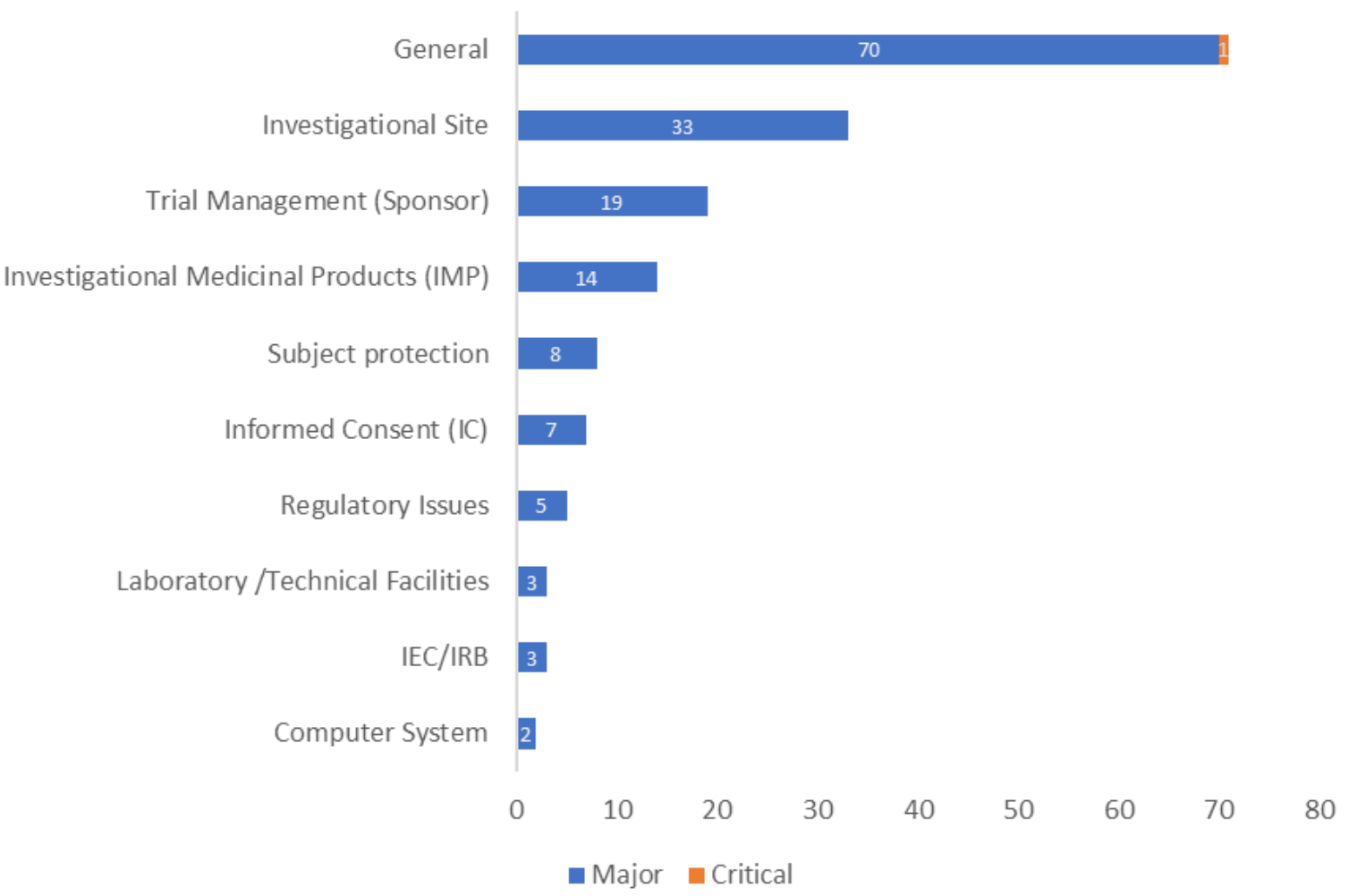
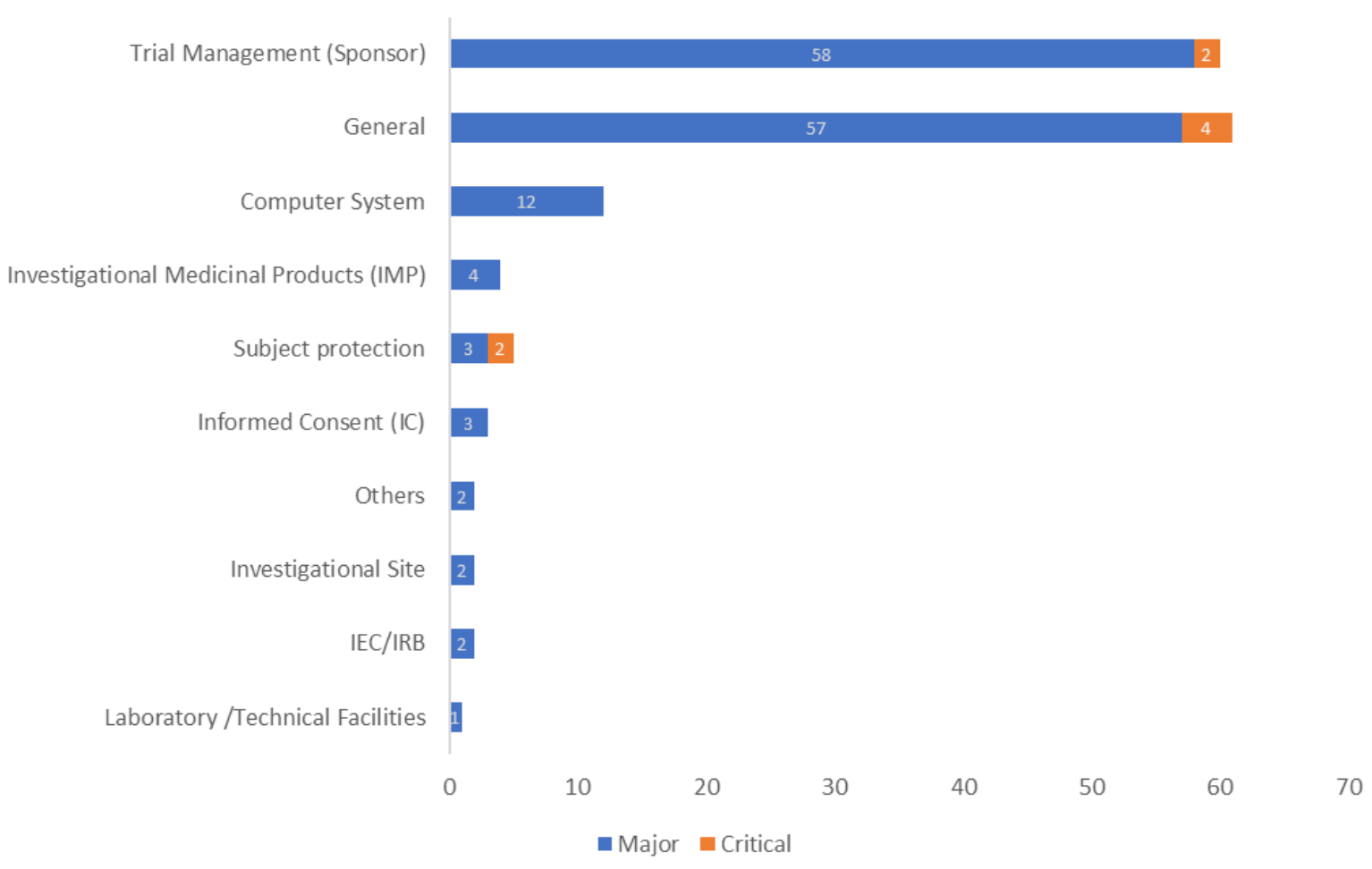
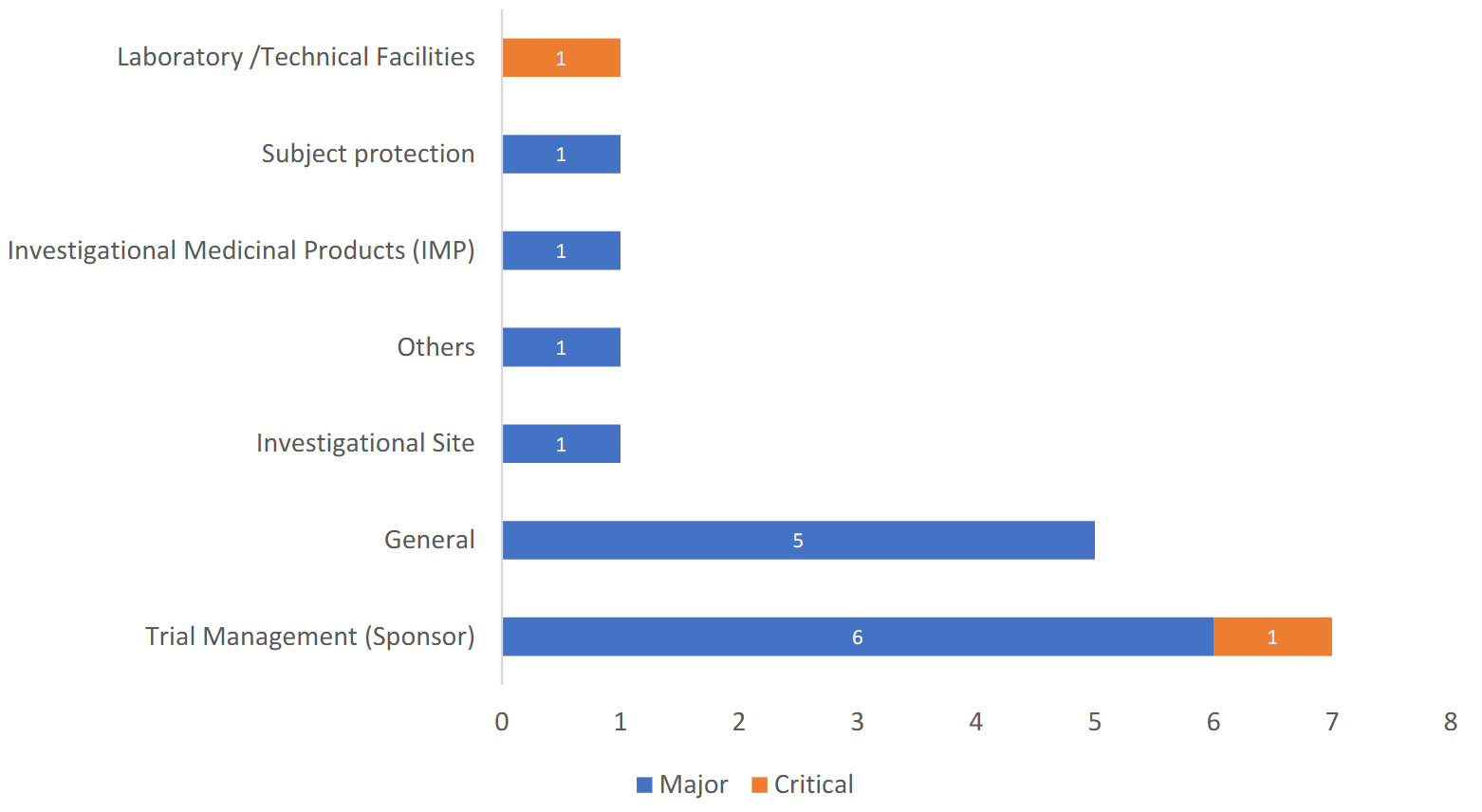
合同/协议中缺乏明确的GCP合规条款(Lack of explicit GCP compliance clauses in contracts/agreements)
对试验文件的直接访问权限不足(Insufficient direct access to trial documentation)
第三方/供应商资质认定与监督记录不充分(Inadequate documentation of third party/vendor qualification and oversight)
合同/协议未正式签署或延迟(Missing or delayed formalisation of contracts/agreements)
申办者与临床研究者的合同/协议未明确文件保存与归档要求(Retention and archiving requirements not established in contracts/agreements between sponsors and clinical investigators)
必备文件和数据直接访问(Essential documents and direct access to data):
监查员和检查员无法在试验现场访问包含关键文件/数据的试验相关电子系统(Lack of access at the trial site to trial-relevant electronic systems containing essential documents/data for monitors and inspectors)
试验主文件/研究者场地文件问题,包括文件不完整或维护不善(Trial Master File/Investigator Site File issues including incomplete or poorly maintained files)
版本控制和文件管理缺失:工作表、知情同意书缺少版本控制;方案与研究者手册内容不一致;试验主文件(TMF)质量控制流程缺乏正式文件记录(Lack of version control and document management: missing version control of worksheets, informed consent forms, inconsistencies between protocol and investigator brochure; no formally documented process for TMF quality control)
复印件认证和质量不足:未建立复印件认证流程;源数据验证/审查仅基于未经认证的电子病历副本(Inadequate certification and quality of copies: Process for certification of copies not established; source data verification/review only based on review of uncertified copies of electronic medical records)
临床试验机构的检查就绪度不足(Lack of inspection readiness at clinical sites)
设施和设备(Facilities and equipment):
存档和存储设施不足(Inadequate archiving and storage facilities)
设备验证、维护与文件记录缺陷:缺少设备校准、诊断设备无规范文件记录、设备维护证书缺失(Equipment qualification, maintenance, and documentation deficiencies: Lack of equipment calibration, diagnostic equipment lacking proper documentation, equipment maintenance certificates not available)
对设施是否适合试验程序缺乏评估或评估不足(Lack of or insufficient assessment of the suitability of facilities for trial procedures)
缺乏生物样本临时存储位置及存储条件的文件记录(Lack of documentation on temporary storage location and the conditions of storage of biological samples)
组织和人员(Organisation and personnel):
试验相关任务授权缺陷(Deficiencies in delegation of trial related tasks):
o 在委托期限外执行任务(Tasks performed outside of the delegation period)
o 记录的任务委托与实际执行活动之间存在差异(Discrepancies between documented delegation of trial related tasks and actual activities performed)
o 申办者/CRO提供的委托日志模板不完善(Inadequate delegation log template provided by the sponsor/CRO)
资质/培训(Qualification/training):
缺失或不完整的培训文件(如资质证明、培训记录、GCP培训证书等),无法证明研究中心人员接受了GCP及试验相关文件的培训(Missing or incomplete training documentation (e.g. qualifications, training records, GCP training certificates, etc.) evidencing site personnel training in GCP and trial relevant documents)
现场人员培训延迟或时机不对(如在试验启动或方案实施后才完成培训)(Delayed or untimely training of site personnel (training completed after site activation, training completed after protocol implementation) )
盲法相关文件不完整或缺失,和/或随机化/盲法变更的沟通不当(Inadequate or missing blinding-related documentation and/or inappropriate communication of changes to randomisation/blinding)
CSR中对方案偏离和关键数据的报告不完整或不准确(Incomplete or inaccurate reporting of protocol deviations and key data in the CSR)
数据纳入和分析的延迟与缺陷:数据清洗不足,原因是主要分析后才检测到数据变更(Delays and gaps in data inclusion and analysis: Insufficient data cleaning resulting in data changes detected only after primary analysis)
数据管理(Data Management):
缺乏数据管理计划、程序与监督(Lack of data management plans, procedures, and oversight)
数据迁移与系统验证不充分(Inadequate data migration and system validation)
数据录入、清洗和锁定的延迟与缺陷(Delays and gaps in data entry, cleaning, and freezing)
数据处理的记录与可追溯性不足:使用Excel收集敏感数据;方案偏离的识别与报告流程不完善(Inadequate documentation and traceability of data handling: Use of excel for sensitive data collection; insufficient process for identifying and reporting protocol deviations)
数据集与分析结果的版本控制及时间戳不充分(Inadequate version control and date-time stamping of data sets and analysis outcomes)
数据安全性不足:伪匿名化和揭盲数据未采用密码保护或加密传输(Inadequate data security: Pseudonymised and unblinded data sent without password protection or encryption)
文件控制(Document control):
缺乏正式的文件审核与控制流程(Lack of formal processes for document review and control)
临床试验关键决策文件记录不完整或不一致(Incomplete or inconsistent documentation of key decisions in the clinical trial)
监查(Monitoring):
监查拜访与相关文件记录不充分或延迟(Inadequate or delayed monitoring visits and documentation)
监查工作未基于适当的监查计划(Monitoring not based on an appropriate monitoring plan)
问题发现与上报机制不足:严重不良事件(SAE)报告延迟、研究场地文件(ISF)文件缺失、方案偏离、实验室数值遗漏等问题未被及时发现(Inadequate detection and escalation of issues: delays in SAE reporting, missing ISF documents, protocol deviations, missing laboratory values not detected)
监查员对源数据的访问权限不足:临床监查员(CRA)无法直接访问电子健康档案(EHR)/电子病历(EMR),限制了其执行监查任务的能力;并非所有可用源数据均可被监查(Insufficient access to source data for monitors: CRAs did not have direct access to EHRs/EMRs, limiting their ability to perform monitoring tasks; not all available source data used for monitoring.)
方案及相关文件的错误与歧义:编辑错误、表格标题误导、方案中未规定非计划访视的说明(Errors and ambiguities in protocol and associated documents: editorial errors, misleading table titles, no instructions in the protocol for unscheduled visits)
方案修订的文件记录存在延迟与缺陷(Delays and gaps in documenting protocol amendments)
病例报告表与日记的设计与实施不完善(Inadequate design and implementation of CRFs and diaries)
入选排除标准及患者数据的记录不完整或前后不一致(Incomplete or inconsistent documentation of inclusion and exclusion criteria and patient data)
统计分析(Statistical Analysis):
最终分析数据集的处理与存储不当(Inadequate handling and storage of final analysis data sets)
数据清洗与分析工作存在延迟与缺陷(Delays and gaps in data cleaning and analysis)
统计报告不完整或不准确(Incomplete or inaccurate statistical reporting)
统计分析程序变更的依据缺乏文档记录(Lack of documentation of rationale for changes in statistical analysis procedures)
研究现场(Investigational site)缺陷
方案依从性(其它)(Protocol compliance (others)):
未发现和未报告的方案偏离:(Undetected and unreported protocol deviations:)
o 与IMP给药时间相关的方案偏离(Protocol deviations related to the timing of IMP administration)
o 方案偏离的发现和记录延迟(Late detection and documentation of protocol deviations)
与受试者安全相关的方案偏离:(Protocol deviations relevant to participant safety)
o 在受试者随机分组前未完成资格评估(Eligibility assessment was not completed before the randomisation of participants)
与临床样本管理不当相关的方案偏离(Protocol deviations related to improper management of clinical samples)
方案偏离以及在未修改方案下变更试验操作(Protocol deviations and changes to trial conduct without a protocol amendment)
方案依从性(疗效评估)(Protocol compliance (assessment of efficacy)):
未执行或未妥善记录研究方案要求的所有疗效评估:缺失实验室检查、未完成全部临床评价、未按方案规定的顺序执行评估和问卷(Failure to perform or properly document all efficacy assessments required in the study protocol: Missing laboratory tests, failure to conduct all clinical evaluations, not following the protocol-specific order for assessments and questionnaires)
研究现场安全性报告流程存在缺陷,导致AE及SAE的报告遗漏或不完整(Deficiencies in the safety reporting process at the site leading to under- or incomplete reporting of AEs and SAEs)
SAE报告缺乏研究者监督:缺少主要研究者对SAE的审查和签字确认证据(Lack of investigator oversight of SAE reporting: Lack of evidence of principal investigator review and sign-off on SAEs)
研究现场在安全性事件报告方面的培训和程序不完善(Inadequate training and procedures related to safety event reporting at the investigational site)