|
首页
>
资讯
>
日本 PMDA 实施新版药品 GMP 检查指南
出自识林
日本 PMDA 实施新版药品 GMP 检查指南
2026-04-15
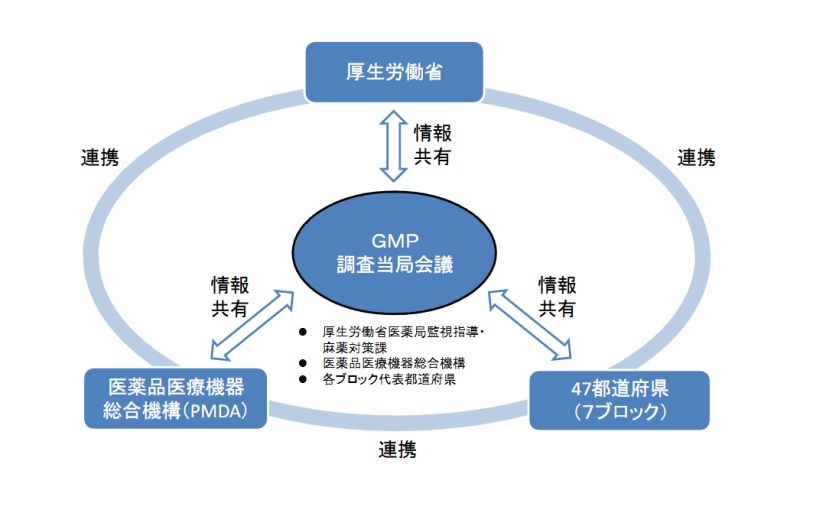
3月19日,日本厚生劳动省(MHLW)定稿发布修订版药品GMP检查指南(GMP 調査要領の制定について)。该文件系统规定了药品生产企业GMP符合性检查的管理框架与操作规范,内容详尽,可供我国出海药企全面了解和准备日本药品和医疗器械管理局(PMDA)检查。
篇幅所限,本文仅就部分重点内容做概要导读。识林会员也可登录查阅日本PMDA发布的“橙色警告信”。
检查分类和频次:针对变更管理的检查,高风险每年飞检
PMDA将检查分为五类:
其中第3类可以理解为一种基于产品类别(如无菌、非无菌等)的监管机制:只要工厂在有效期内通过了该类别的GMP检查,同类别的新产品通常可简化或豁免现场检查。
第4类也比较特别,又细分为包括已上市产品变更管理中的合规确认以及境外特例批准产品变更管理中的合规确认。
检查指南第9.1条明确了检查频次。PMDA针对每个生产场地实施基于风险评估的检查,检查周期为1至3年。原则上以现场检查为主,但根据风险评估结果也可采用书面检查。针对高风险生产场地,原则上每年实施1次以上的飞行检查(無通告立入検査)。
PMDA通常预先向被检查单位告知检查日期、检查员人数、更衣尺码等必要信息。对于境外生产企业检查,若已选定境内代理人,PMDA需事先联系该境内代理人,并指示其向被检查生产企业传达。对于飞行检查,若有其他人员(如下文提到的观察员)作为检查组成员加入,PMDA需在检查开始前获得被检查生产企业的同意。
检查人员组成:建议2名以上,观察员需企业认可
PMDA原则上需确保1名以上检查员,并根据需要纳入相关领域的专家,组成检查组。从专业性与经验的相互补充及检查员安全的角度,通常建议检查组由2名以上成员组成。检查组长负责检查实施的整体管理、现场讲评、缺陷项目的传达以及检查报告的编制。
对于观察员,在开始检查之前需获得被检查生产场地所属的生产企业等及检查组长的认可。观察员包括互认协议(MRA)国家药品监管部门的职员及相关机构的职员。PMDA需向观察员指示遵守保密义务等必要事项,观察员需遵从该指示。
三类缺陷:严重缺陷重点关注于产品危害或欺骗篡改
检查缺陷按以下基本思路分类。
严重缺陷需同时满足两个条件。首先,该缺陷违反了GMP规范条款;其次,满足以下任一情形:产品对患者有害,或存在制造有害产品的明确风险;生产企业对产品或记录实施了欺骗、虚假报告或篡改。
主要缺陷指违反了GMP规范条款,但不属于严重缺陷的情形。
一般缺陷指虽不能明确认定违反了GMP规范条款但需改善的事项,旨在进一步完善生产质量及风险管理。
三种结论:严重缺陷15日内整改到“妥当”,仅召回还不够
无缺陷项时或仅存在一般缺陷时,可评定为“符合要求”。对于后者,PMDA要求被检查生产企业等提交整改报告或整改计划。下次常规生产现场检查等时需确认整改情况。
存在主要缺陷时,PMDA要求被检查生产企业等自GMP检查缺陷项通知书送达日起30个工作日内,提交详细整改报告或具体整改计划。若PMDA无法判断内容合理,原则上评定为“不符合要求”。若内容合理,可评定为“符合要求”。对于提交整改计划后评定为“符合要求”的情形,需在整改完成后提交整改报告并确认整改情况。一般缺陷按前述规则处理。
存在严重缺陷时,除非自GMP检查缺陷通知书送达日起15个工作日内完成PMDA认为“妥当”的整改(妥当と判断する改善を完了),否则原则上评定为“不符合要求”。主要及一般缺陷分别按上述规则处理。且PMDA明确指出,主动召回并不被视为严重缺陷的整改。
作者:识林-实木
责任编辑:识林-木姜子
识林®版权所有,未经许可不得转载。
【文件概要】
该文件由日本厚生劳动省制定,旨在强化GMP(药品生产质量管理规范)调查体系,统一调查标准并提升监管透明度。文件整合了现行调查流程,新增调查结果报告书的信息收集、积累、分析与共享机制,要求调查当局(包括都道府县及PMDA)建立质量管理监督系统,制定质量手册,明确调查分类(适合性调查与准入检查等)、实施程序及结果公布流程。重点内容包括:调查频度基于风险评估(高风险制造所年检1次以上,其他1-3年1次);新增无通告立入检查以防范系统性隐瞒;要求调查团队需包含符合资质的调查员;引入GMP/GCTP质量信息系统实现数据共享;对不配合调查或严重违规的制造所采取行政措施(如许可取消)。文件废止了2022年旧通知,自2026年4月1日起实施。 【适用范围】
本文适用于日本境内所有医药品及医药部外品的制造所(含化学药、生物制品、疫苗等),涵盖创新药、仿制药、原料药及出口专用药品。调查当局包括47个都道府县及PMDA,企业类型涉及大型药企、跨国药企、Biotech及CDMO。不直接适用于医疗器械或化妆品领域。 【影响评估】
本文要求企业强化内部GMP合规体系,应对更频繁的无预警检查及数据披露义务。违规成本显著增加,例如严重不合规将导致调查结果公开及许可取消风险。企业需投入资源完善文档管理、变更控制及员工培训,以符合新增的稳定性监测、风险管理和供应商审计要求。跨国企业需注意与PIC/S等国际标准的协调。 【实施建议】 - QA/QC:必读。修订质量手册,强化偏差管理及CAPA系统,确保符合新增的稳定性监测要求。
- 生产:必读。优化清洁验证及工艺验证流程,应对无预警检查中的现场数据审查。
- 注册:必读。跟踪调查结果报告书提交时限(10个工作日内),协调跨部门响应监管问询。
- 供应链:评估原料供应商审计标准,确保符合文件新增的供应商管理条款。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |