|
首页
>
资讯
>
FDA 定稿三篇关于抗癌药开发临床试验的重要指南
出自识林
2022-03-02
美国 FDA 于 2022 年 3 月 1 日发布了三篇关于抗癌药开发的定稿指南,重点关注两个重要概念:主方案(master protocols)和扩增队列(expansion cohorts),并强调了癌症临床试验应如何包括 75 岁以上的参与者。
FDA 在新闻稿中表示,这三篇定稿指南的发布是为了配合美国总统拜登重新推动他的癌症登月计划。该计划最初由拜登于 2016 年担任副总统期间发起,2022 年 2 月这项工作重新被提起。在新的癌症登月计划下,拜登政府的目标是在未来 25 年内将癌症死亡率降低 50%,并改善癌症患者及其家人的生活和癌症生存体验。
FDA 肿瘤学卓越中心主任 Richard Pazdur 表示,“通过今天的行动,FDA 正在推荐一些重要原则,以解决不平等问题、针对正确的患者提供正确的治疗、加快针对最致命和罕见癌症的开发进展,并从所有患者体验中学习。所有这些都是癌症登月计划的宗旨。”
主方案:加速肿瘤药和生物制品开发的高效临床试验设计策略
这份 24 页的主方案指南主要基于 2018 年 10 月的草案版本,解释了如何开展这些试验,以在同一试验架构内同时评估一种以上的研究药物和/或一种以上的癌症类型。临床试验主方案设计一直以来是 FDA 鼓励的方向,现 FDA 首席副局长 Janet Woodcock 长期是主方案的坚定支持者。
主方案通常分为两种形式:“篮式试验” — 旨在测试一种研究药物或药物组合用于不同的癌症;“伞式试验” — 旨在评估单一疾病人群中的多个不同研究药物。
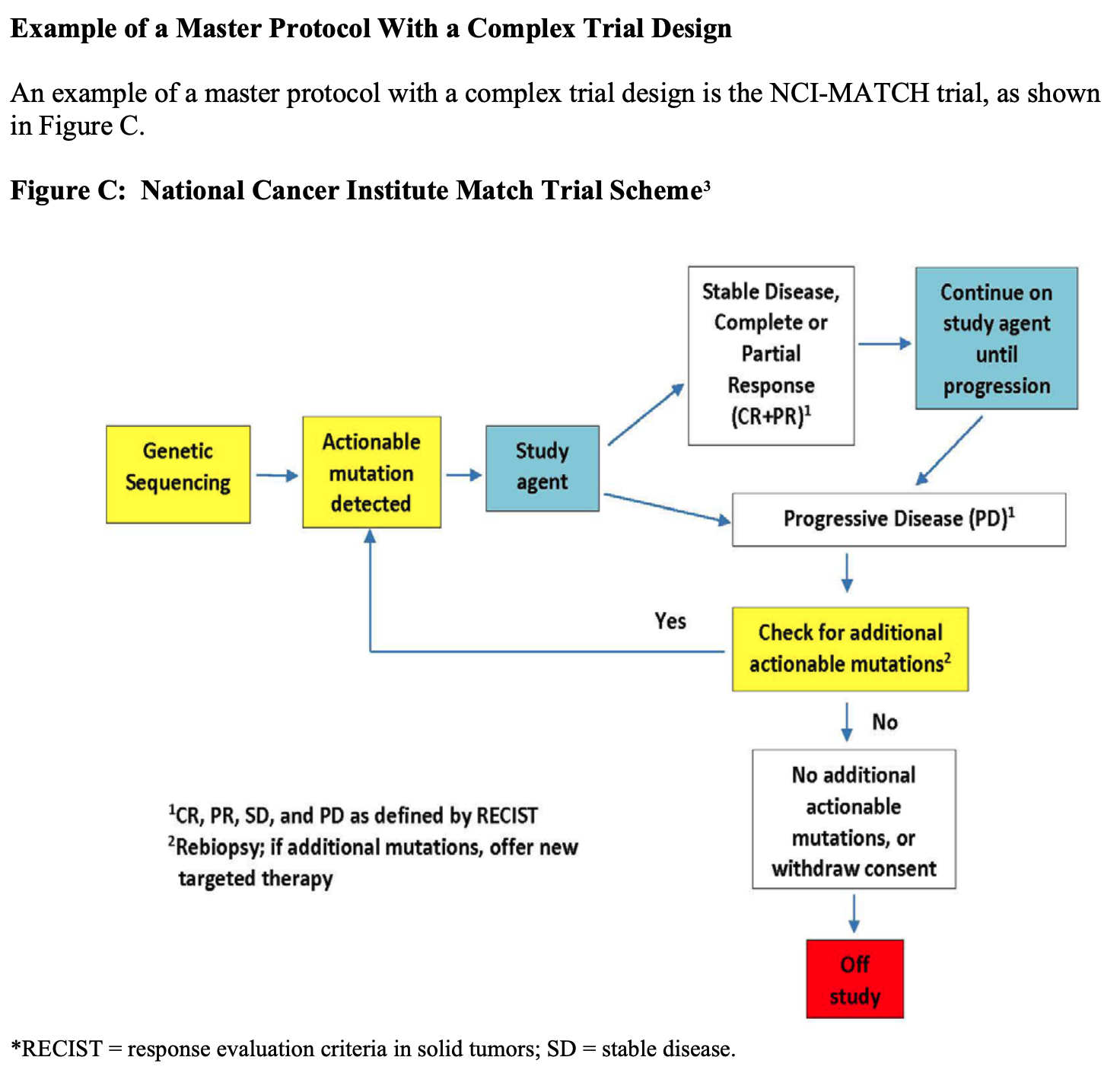
FDA 指出主方案设计还可以结合篮式试验和伞式试验的设计特征,提供了 NCI-MATCH 试验(试验框架见下图)作为具有复杂试验设计的主方案的示例,该试验试图确定无论癌症类型如何,根据特定基因变化治疗癌症是否是有效的。
定稿指南“对指南草案的各个部分进行了修订,以澄清需要提交给 FDA 以支持扩大方案的增补、累积安全性更新的频率、特设机构审查委员会会议的作用、安全评估委员会的作用以及知情同意要求等方面的信息。”
扩展队列研究:在人体首次临床试验中使用以加速肿瘤药物和生物制品的开发
在这篇 16 页的定稿指南中,FDA 举了两个例子,分别是 KEYNOTE-001 试验(在晚期非小细胞肺癌患者中研究了默沙东重磅抗癌药 pembrolizumab)和 EMD Serono 的 JAVELIN 试验(研究了其抗癌药 Bavencio)。
首次人体(FIH)多重扩展队列试验旨在通过从初始确定耐受剂量到评估更典型的 2 期试验(即估计抗肿瘤活性)在单个扩展队列中的无缝进行来加快开发。一般来说,这些扩展队列将在研究药物的代谢和药代动力学分析之前启动,安全性评估有限。因此,鉴于此类试验已招募了数百至 1,000 多名受试者,因此确保在扩大的队列中对毒性进行密切和及时的监测至关重要。
FDA 还指出了从 2018 年 8 月的指南草案到定稿指南的几个“重大变化”,包括添加“除西蒙两阶段设计之外的设计(例如,贝叶斯统计设计)可用于评估非随机队列中的抗肿瘤活性以限制可能接触潜在无效药物的受试者数量”的文字内容。
另外 FDA 还添加了一项声明“以表明在有限样本量的试验中,安全评估委员会可以是申办者组织内的一个单独团体,不参与试验执行或管理,或者是由外部代表代替独立的数据监测委员会。”这是行业喜闻乐见的规定。
FDA 还对各个部分进行了微小修订,以阐明适用于具有 FIH 多重扩展队列的临床试验的药物标准,在患者管理时使用体外诊断时获取风险评估的程序,以及需要提交给 FDA 以支持扩展方案增补的信息。
在癌症临床试验中包括老年人
这份指南是 FDA 银发计划(Project Silver)的一部分,该计划是 FDA 肿瘤卓越中心的一项公共卫生计划,旨在增加老年人(65 岁及以上)在癌症临床试验中的代表性。
据 FDA 称,从 2010 年到 2030 年,65 岁及以上人群的癌症发病率预计将增加 67%,FDA 正试图鼓励行业将更多这些老年人纳入试验。 FDA 表示,最终指南强调了将 75 岁以上的成年人纳入癌症临床试验的特殊重要性。
作者:识林-Aspen
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位: - 临床(Clin):必读。应深入理解文件内容,以确保临床试验的设计和执行符合FDA的最新指南,特别是在涉及多个扩展队列时。
- 注册(Reg):必读。需熟悉指南以准备和更新IND申请,确保与FDA的沟通和协议符合规定。
- 研发(R&D):必读。在药物开发过程中,应考虑指南中提到的药物特性和临床试验设计,以优化开发计划。
- QA:必读。确保临床试验的质量和合规性,遵循FDA的指导原则。
工作建议: - 临床(Clin):在设计和执行FIH临床试验时,特别注意多个扩展队列的安全性和药效评估,以及数据的实时评估和传播。
- 注册(Reg):在准备IND时,包括有关多个扩展队列的所有相关信息,并与FDA沟通以获得对计划的认同。
- 研发(R&D):在药物开发早期阶段,考虑药物特性是否适合进行多个扩展队列试验,并规划相应的临床试验设计。
- QA:监督临床试验的执行,确保所有活动符合FDA指南,特别是在保护受试者和数据管理方面。
适用范围:
本文适用于肿瘤药物和生物制品的人体首次临床试验,特别针对化学药和生物制品,包括创新药和生物类似药。适用于在美国进行注册的大型药企、Biotech公司以及跨国药企。 要点总结:
FDA的指南强调了在首次人体临床试验中使用多个扩展队列的研究设计,以加速肿瘤药物和生物制品的开发。这些设计允许同时进行多个受试者队列,以评估药物的安全性、药代动力学和抗肿瘤活性。指南详细讨论了适用的药物特性、IND申请中的信息要求、与FDA的互动时机以及保护受试者的措施。特别强调了在设计和实施这些试验时需要考虑的挑战和风险,包括及时传播新的安全信息、避免使受试者暴露于可能的次优或有毒剂量,以及可能对初步试验结果的误解。此外,指南还提供了关于统计考虑、安全性监控、IRB/独立伦理委员会的作用和知情同意文件的更新的具体指导。 以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - 临床(Clin):必读。负责理解和执行主方案中的临床试验设计,确保试验的顺利进行,并及时与FDA沟通。
- 注册(Reg):必读。需要熟悉主方案的要求,以便在IND提交和药物上市申请中正确应用。
- 研发(R&D):必读。在药物开发过程中,需要考虑主方案的设计和执行,以加速药物研发。
工作建议: - 临床(Clin):在设计和执行主方案时,特别注意共同控制臂的使用、多个生物标志物的研究以及独立数据监测委员会(IDMC)的角色和职责。
- 注册(Reg):在IND提交中,确保包含所有必要的主方案元素,并与FDA沟通以获得对设计和执行的反馈。
- 研发(R&D):在药物组合和生物标志物开发计划中,与FDA早期沟通,确保科学合理性,并在试验中使用经过分析验证的生物标志物测试。
适用范围:
本文适用于肿瘤药物和生物制品的研发,特别关注于成人和儿童癌症的治疗。它涵盖了创新药和生物类似药的临床试验设计,包括单臂和多臂试验,以及适应性/贝叶斯设计。适用于美国FDA监管下的Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。 要点总结:
本文强调了主方案(Master Protocols)在肿瘤药物和生物制品研发中的重要性,特别是在评估多种药物和/或多种癌症亚型时的高效临床试验设计。主方案允许在单一试验结构内同时评估多个研究药物和/或癌症类型,提高了药物开发的灵活性和效率。文件中特别强调了共同控制臂的使用、多种生物标志物靶向药物的研究、以及在试验中增加或停止治疗臂的条件。此外,还讨论了生物标志物开发、统计考虑、安全性监测和报告计划、以及与FDA的沟通和互动。主方案的设计和执行需要确保受试者安全,并生成符合监管标准的安全和有效性数据。以上仅为部分要点,请阅读原文,深入理解监管要求。 |