首页
>
资讯
>
FDA 鼓励临床试验采用主方案设计
出自识林
2019-03-05
美国《21 世纪医药法案 》带来的创新监管方法正在使新药研发现代化。具有主方案(Master Protocol)的临床试验 设计是加速肿瘤药和生物药 研发的现代化方法的一个例子。由于这些试验的复杂性和潜在的监管影响,这些试验经过良好设计并得到很好的执行对于确保患者安全性和获得可支持药品批准的质量数据是重要的。
传统上,肿瘤药研发涉及一系列临床试验,研究单一疾病中的一个或两个药。另一类临床试验(即,具有主方案的临床试验)则旨在同时评估在成人和儿科癌症中相同整体试验结构内的一种以上研究用药物和/或一种以上癌症类型。《主方案 — 加速癌症药和生物药研发的高效临床试验设计策略 》行业指南草案为后一类临床试验的设计和执行提供了建议。主方案可包含需要特殊考虑的具体试验特点。主方案设计类型的示例包括通常称为篮式和伞式的试验:
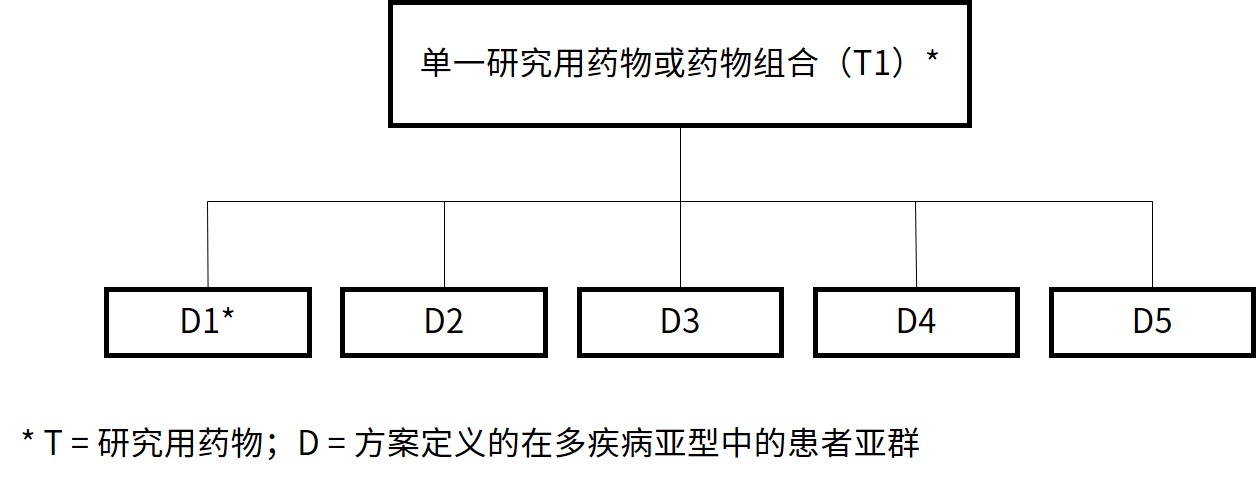
篮式试验 涉及在多个癌症群体中研究单一研究用药物或药物组合,癌症群体由疾病阶段、组织学、先前所用治疗药物数量、遗传或其它生物标志物 ,或人口统计学特征定义。篮式试验通常被设计为单臂,以总体应答率作为主要终点的活性评估试验。在子研究中看到的强烈应答信号可允许扩展以生成可支持上市批准的数据。
图 1:篮式试验设计主方案示意图
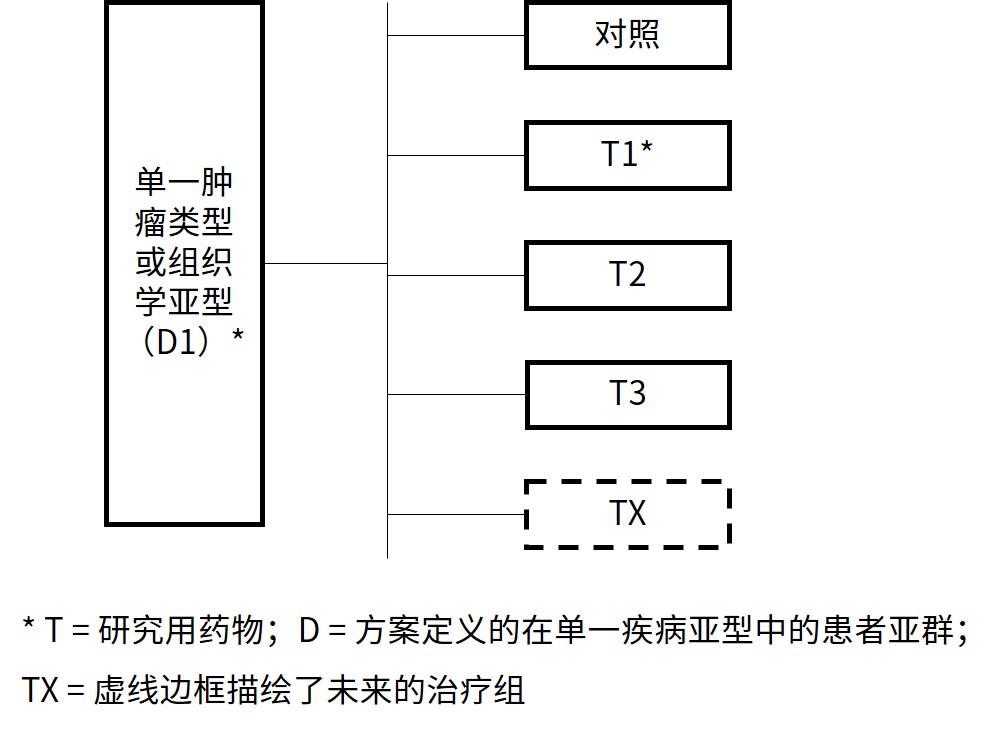
伞式试验 旨在评估单一疾病人群中多个研究用药物作为单一药物或组合药物用药。子研究可以包括剂量确定部分,以在开展活性评估部分之前确定研究用药物组合的安全剂量。
图 2:伞式试验设计主方案示意图
主方案中的设计考量点:
给药方案(Dosing) :FDA 强烈建议在主方案评估之前建立每个研究用药的推荐 2 期剂量(RP2D)。
共用对照组(Common Control Arm) :在同时评估针对单一疾病的多种药物的试验中,申办人 可以使用共用对照组,共用对照组应该是目标人群目前的治疗标准(standard of care,SOC)。共用对照组可能会随时间而改变,因为更新的药物有可能替代 SOC。应在试验药物和共用对照之间开展比较分析,而不是在实验性治疗组之间开展。
新型组合(Novel Combinations) :申办人应为旨在评估两个或多个研究用药物同时给药的研究提供强有力的科学依据。申办人还应确保已为每个可能具有抗肿瘤活性的药物确定了 RP2D。
添加和停止治疗组(Adding and Stopping Treatment Arms) :主方案和统计分析计划应根据预先指定的中期分析或外部新数据的结果描述添加、扩展或中止治疗组的条件。
独立的数据监测委员会(Independent Data Monitoring Committee) :如果申办人预计一个或多个子研究的结果将构成上市申请的基础,主方案应描述并提供独立的放射学审查委员会章程,以执行基于肿瘤的盲法评估,以及独立数据监测委员会的章程,以监测有效性结果。
生物标志物开发(Biomarker Development) :评估生物标志物定义的人群的主方案应解释为何使用的生物标志物是合适的,并采用经过分析验证的体外诊断测试。有兴趣开发特定生物标志物测试以作为伴随诊断器械上市的申办人应咨询负责审评体外诊断测试的相应 FDA 中心。
指南文件还描述了主方案设计的其它方面;试验开展;以及相关考量,例如生物标志物的共同开发、统计分析、安全考虑以及主方案内容。提供了有关申办人如何与 FDA 互动以促进高效审评的建议。还讨论了主方案实施和分析方面的挑战,例如评估研究用药迅速出现的安全性问题。
FDA 强烈建议申办人在研发计划早期与临床审评部门讨论他们根据主方案研发药物的计划,以便在提交前获得有关此类方案设计的反馈。FDA 鼓励申办人仔细阅读指南文件。
Renu Lal,药学博士
CDER 小企业协助
译自 FDA 小企业文章“FDA Modernizes Clinical Trials with Master Protocols ”。
【相关阅读】FDA 在新英格兰医学杂志上发表关于临床试验主方案的述评
编译:识林-椒® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
岗位必读建议:
研发(R&D):了解加速药品开发流程的新规定,确保研发项目符合新法规要求。 注册(Regulatory Affairs):掌握法规对药品注册流程的影响,优化注册策略。 临床(Clinical):关注临床试验设计的新指导原则,确保试验合规性。 文件适用范围:
文件要点总结:
加速药品开发流程 :强调了加速药品从发现到上市的整个流程,以促进21世纪医疗创新。临床试验现代化 :提出了对临床试验设计的现代化要求,以提高试验效率和患者参与度。个性化医疗推进 :鼓励发展个性化医疗方法,包括精准医疗和基因疗法。数据共享与隐私保护 :规定了数据共享机制,同时强调了患者数据的隐私保护。监管框架更新 :明确了对FDA监管框架的更新,以适应新兴医疗技术和产品。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
临床研究(Clinical) :必读。负责设计和执行临床试验,确保试验符合指南要求。注册事务(Regulatory Affairs) :必读。负责确保提交给FDA的文件符合指南要求。药物警戒(PV) :必读。负责监测临床试验中的安全性问题。医学写作(Medical Writing) :必读。负责撰写和更新临床试验方案和知情同意文件。质量保证(QA) :必读。负责监督临床试验的质量控制和合规性。工作建议:
临床研究 :在设计临床试验时,考虑使用单一共同对照组以提高效率。注册事务 :在提交IND时,确保包含所有必要的元素,如核心元素、电子提交格式等。药物警戒 :建立系统性方法,确保严重安全问题能够迅速传达给临床研究者和监管机构。医学写作 :在知情同意文件中包含所有临床重要的方案修改和新的安全信息。质量保证 :定期评估临床试验的安全性,确保试验按照IND中的一般调查计划和方案进行。适用范围:
要点总结:
主方案定义 :主方案是设计有多个子研究的方案,可以在整体试验结构中评估一个或多个研究药物在一个或多个疾病亚型中的效果。设计考虑 :使用单一共同对照组可以提高效率,但需要确保对照组是当前标准治疗。生物标志物开发 :在试验中使用生物标志物时,需要确保使用的体外诊断测试具有分析验证。统计考虑 :非随机化方案中,计划的样本量应足以排除临床上不重要的反应率。安全性考虑 :需要建立系统性方法,确保严重安全问题能够迅速传达给临床研究者和监管机构。以上仅为部分要点,请阅读原文,深入理解监管要求。