|
首页
>
资讯
>
FDA 应用监管科学处聚焦新方法学(NAM)替代动物试验
出自识林
FDA 应用监管科学处聚焦新方法学(NAM)替代动物试验
2026-03-04
2月25日,FDA发布《应用监管科学处2025年度报告》,作为隶属于转化科学办公室(OTS)临床药理学办公室(OCP)的核心科学部门,应用监管科学处(DARS)在2025年度聚焦于新方法学(NAM)的开发与应用,在心脏/肺部NAM模型、罕见变异体及药物组合评估等领域取得系列进展,并为药品审评提供了高频次的技术咨询。
以2025年FDA发指南简化单克隆抗体非临床研究为里程碑,NAM日益成为全球药品监管科学焦点。这份报告显示出基于细胞、组织或计算机模拟的NAM已在心脏毒性评估、肺渗透性检测、药物相互作用及罕见代谢变异体研究等多个领域从概念走向应用。创新药企在IND和NDA申报中主动引入经充分验证的NAM数据(尤其是利用官方文献中提及的工具),或成为加速审评、降低开发成本的新路径。同时,NAM的验证与标准化本身即构成技术壁垒,抢先建立内部NAM能力的企业有望在未来监管对话中占据主动。
DARS聚焦于NAM领域
报告中涉及药物的研究课题,大部分都与NAM有关。摘要如下:
*如需具体文献标题和链接,可参阅报告。
已完成课题:
DARS与新药办公室(OND)合作开展的人类心脏NAM研究旨在减少药物安全性评估对动物试验的依赖。DARS科学家通过严格的模型表征和实验方案开发,证实人类心脏模型能够成功复现临床结局,准确检测心脏安全性问题。该研究同时建立了全面的良好操作规范和使用场景(context-of-use)建议。
另一项已完成课题是与仿制药办公室(OGD)合作建立的肺部NAM用于药物渗透性评估。该研究聚焦吸入性药物的渗透性测量,利用用于慢性阻塞性肺疾病(COPD)和哮喘治疗的支气管扩张剂成功完成模型表征。该模型可为监管人员提供更精确的渗透性评估数据,支持吸入性药物的审评决策。
在研课题:
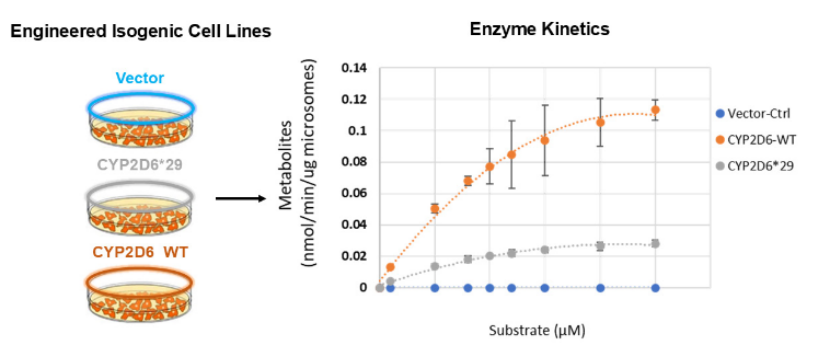
在研课题“验证用于表征罕见药物代谢变异体的体外细胞模型”取得阶段性进展。为阐明代谢酶罕见遗传变异体的影响,DARS与转化与精准医学处(DTPM)合作,构建了工程化HEK293细胞模型,用于比较某个临床意义明确的基因变异型与同一基因常见型的药物代谢能力。初步研究表明,CYP2D6*29变异型酶的药物代谢活性较常见型显著降低。该结果与临床数据一致,证实这些细胞模型能够反映具有临床意义的活性阈值。未来研究人员可更有信心地利用这些模型预测尚未充分研究的基因变异体对药物代谢的影响,包括理解临床意义尚不明确的遗传变异。
新课题:
2025年新启动的课题中,DARS开展心脏NAM评估药物联合应用研究。针对患者同时使用多种药物的临床现实,DARS利用新型心脏NAM研究了考比司他(cobicistat)与莫西沙星(moxifloxacin)的联合应用,发现考比司他可降低莫西沙星诱导的心脏复极化延长风险。该研究既展示了药物相互作用可产生保护效应,也提示该检测系统可用于识别可能加重心脏风险的药物组合。
另一项新启动的神经NAM研究响应了FDA于2025年4月发布的《减少临床前安全性研究中动物试验的路线图》的战略方向。DARS牵头利用人类神经NAM应对阿片类药物领域的重大挑战,包括药物滥用、阿片类药物使用障碍、合成阿片类药物问题以及扩大治疗和逆转选择。
其他工作:可视化杂质风险,参与药物和DDT审评,复核(Q)SAR评估
除了上述以NAM为核心的探索性研究,DARS在2025年还承担了大量的日常监管支持工作,为药品审评提供了直接的技术支撑。
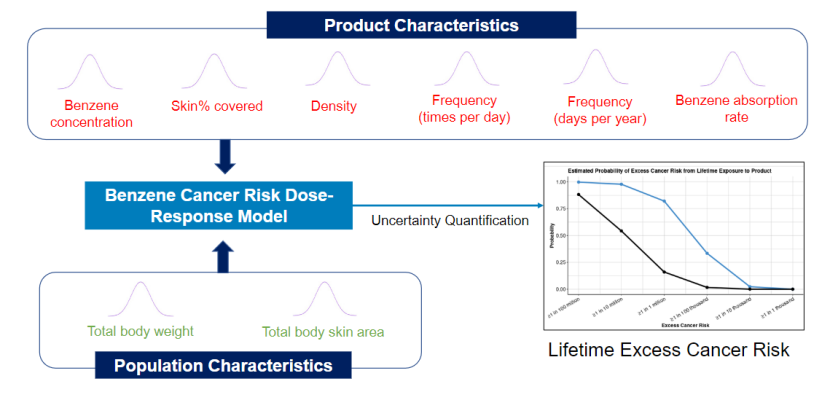
DARS持续开发定量化且用户友好的杂质与污染物风险评估工具。该工具用于评估苯(一种已知致癌物)等杂质和污染物的风险。在实际监管应用中,该工具已用于支持部分外用药品涉嫌苯污染事件的召回分类决策。
DARS在2025年度完成31次内部监管咨询,涉及的问题涵盖作用机制、方法学路径的适宜性以及监管研究结果的解读。其中包括19份 preIND/IND申请咨询和16份NDA/BLA/ANDA 申请咨询。
在药物开发工具(Drug Development Tools, DDT)方面,DARS参与了新药创新科学和技术方法(ISTAND)的工作,其旨在支持药物研发新方法的开发并推动其获得监管认可,并已于2025年7月正式转为常规DDT资格认证计划。2025年,DARS共参与审评了11项独特的DDT申报。
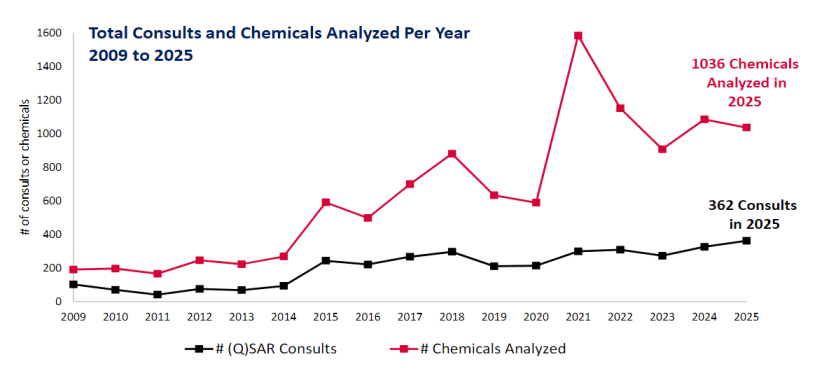
DARS计算毒理学咨询服务的核心工作是为FDA提供基于(定量)构效关系((Q)SAR)分析的内部咨询服务。该服务主要审查制药企业提交的药物杂质(包括亚硝胺类杂质)以及容器密封系统和给药系统中可提取物与浸出物的(Q)SAR分析数据。必要时,DARS内部自行生成(Q)SAR预测结果并进行化学信息学分析,以支持申报资料的审评。2025年全年,DARS累计完成362次(Q)SAR内部咨询,评估了1036个分子。这一数据提示,基于构效关系的计算机模拟分析,已成为仿制药和新药杂质(尤其是亚硝胺)审评中的常规且不可或缺的工具。
识林-实木
识林®版权所有,未经许可不得转载
【文件概要】
该文件提出FDA通过新方法学(NAMs)减少临床前安全性研究中动物试验的战略路线图,涵盖器官芯片、计算建模和先进体外检测等技术。NAMs通过整合人类相关模型(如微生理系统、人工智能预测工具)提高药物安全性预测的准确性,同时降低动物使用。FDA计划优先在单克隆抗体(mAb)领域实施,逐步扩展至其他生物分子和化学实体。文件概述了3年内的实施步骤,包括利用国际数据、建立开放数据库、缩短灵长类动物毒性试验周期,并通过ICCVAM协调跨机构合作以加速NAMs验证。长期目标是将动物试验转为例外,建立以NAMs为核心的新标准。 【适用范围】
本文适用于美国FDA监管的生物制品(如单克隆抗体)、创新药及生物类似药的临床前开发阶段,主要针对大型药企、Biotech公司及CRO/CDMO。 【影响评估】
本文将显著降低药企在非临床研究中的动物试验成本和时间,但需投资NAMs技术验证及数据整合。合规策略需转向多学科协作,以应对监管对NAMs数据要求的提升。 【实施建议】 - 必读岗位:
- 研发(非临床): 评估器官芯片和计算模型替代传统动物试验的可行性,优先在mAb项目中试点。
- 注册: 跟踪FDA对NAMs的接受标准,在IND/BLA提交中整合支持性NAMs数据。
- QA: 建立NAMs实验的GLP等效标准,确保数据可追溯性。
- 建议行动: 参与FDA试点项目,与ICCVAM合作验证方法,定期审查动物试验替代方案的经济性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |