首页
>
资讯
>
FDA 指导 Ames 阳性临床药物的后续测试
出自识林
2024-12-02
美国 FDA 于 11 月 26 日发布了《药物(活性成分)或代谢物 Ames 阳性情况下为支持健康受试者中首次人体临床试验而建议进行的后续测试》 指南草案,详细说明了其关于如何以及何时可以对具有致突变潜力的药物进行首次人体试验(FIH)的想法。
如果之前的测试显示对健康试验对象的风险很小,FDA 将采取基于风险的方法,允许开发某些致突变产品。FDA 建议申办人 对 Ames 阳性药物活性成分进行后续体外和体内致突变试验。
FDA 将基因毒性 测试定义为体外和体内测试,旨在识别诱导基因损伤(致突变性或致染色体断裂性)的化合物。这些测试可以帮助利益相关者了解药物造成 DNA 损伤及其固定的可能性。
FDA 表示,“基因毒性测试在药物开发的研究性新药申请(IND) 阶段对保护临床试验 对象免受潜在增加的基因毒性危害和癌症风险起着重要作用。通过 ICH 咨询程序,行业和监管机构已经接受了一系列标准的遗传毒理学 研究。”
FDA 列出了几篇 ICH 指南,包括 ICH M3(R2) ,并建议进行致突变性测试以保护试验对象,这些测试通常在 I 期试验开始前或至少 II 期试验之前进行。通常,大多数被发现具有致突变性的药物不会进一步开发以获得 FDA 批准。然而,某些药物,由于其治疗作用机制,尽管具有致突变潜力,还是可能会被 FDA 允许进一步开发。
指南指出,“指南就 Ames 阳性药物活性成分的后续测试提出了建议,以应对申办人决定继续开发的罕见情况。这些建议旨在在健康人类受试者 中进行 FIH 试验之前解决和降低某些安全问题。”
“后续测试不能完全缓解 Ames 阳性结果引起的担忧,而且在缺乏充分的致癌性评估的情况下,仍存在一些残留风险。因此,进一步开发的 Ames 阳性药物活性成分应该是针对严重或危及生命的疾病且医疗需求未得到满足的情况。”
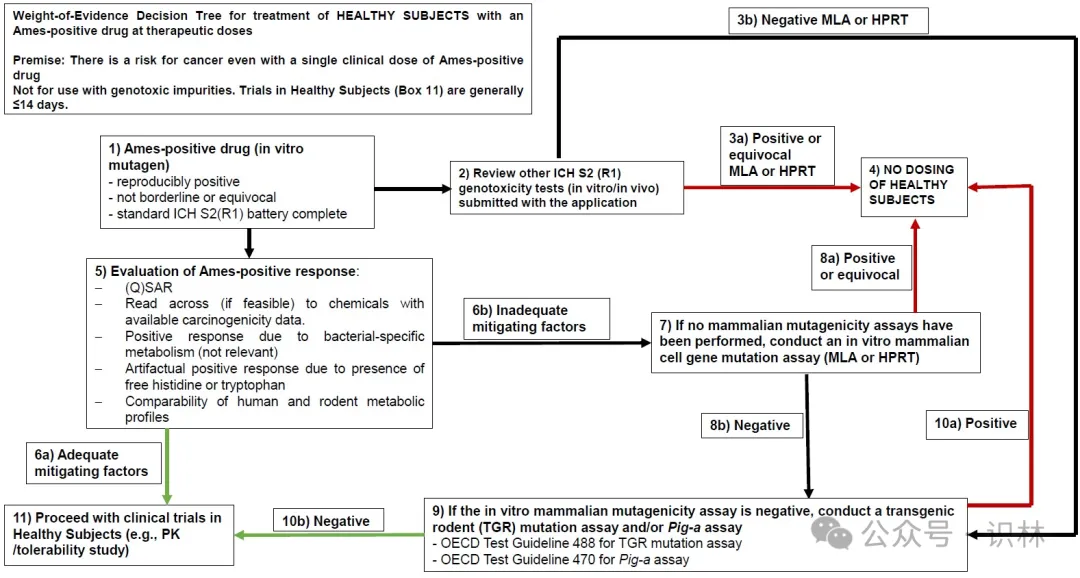
指南指导申办人应如何分析他们的 Ames 测试数据,并在测试结果为阳性时进行后续测试。指南还详细介绍了对 Ames 阳性药物或代谢物进行证据权重 (WoE) 评估。FDA 绘制了一棵决策树,申办人在考虑对健康试验对象使用 Ames 阳性产品时进行 WoE 评估时可以使用。
FDA 表示,只有当先前广泛的后续测试结果根据 WoE 评估降低了癌症风险时,才可能考虑允许将 Ames 阳性药物活性成分用于健康受试者。
“体外哺乳动物细胞基因突变试验和体内基因突变试验都需要为阴性,才能考虑对健康受试者进行 FIH 试验。如果体外哺乳动物细胞基因突变试验或体内基因突变试验为阳性,则不支持对健康受试者进行 FIH 试验。”
指南建议,希望在健康临床试验受试者中使用 Ames 阳性产品的申办人应通过 FDA 药品评估和研究中心 (CDER) 的 IND 前流程与相应部门的审评人员沟通。该指南适用于小分子药物活性成分的开发,生物制品 、晚期癌症患者用药和 DNA 反应性(致突变性)杂质 不在本指南的范围内。
识林-椒
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
临床(Clinical) :必读。在设计首次人体临床试验时,需考虑Ames阳性结果对健康受试者的风险评估,并依据本指南进行后续试验设计。研发(R&D) :必读。在药物活性成分或代谢物的Ames测试呈阳性时,需依据本指南进行后续的体外和体内突变性测试。注册(Regulatory Affairs) :必读。负责确保药物开发和注册过程符合FDA的当前思考,并在IND申请中体现。文件适用范围:
文件要点总结:
Ames阳性后续测试推荐 :对于Ames阳性的药物活性成分或代谢物,提供了后续体外和体内突变性测试的推荐,以支持健康受试者的首次人体临床试验。风险评估与决策树 :强调了使用决策树评估Ames阳性结果的风险,并基于权重证据(WoE)方法决定是否进行临床试验。体外测试 :推荐使用体外小鼠淋巴瘤细胞试验(MLA)或哺乳动物细胞HPRT试验作为Ames阳性的后续测试。体内测试 :对于MLA或HPRT试验阴性的Ames阳性活性成分,推荐进行体内突变性测试,如转基因啮齿动物基因突变试验和/或Pig-a基因突变试验。代谢物测试考量 :对于Ames阳性代谢物,应类似于活性成分进行体外和体内突变性测试,并考虑其在人体中的独特或不成比例的代谢。以上仅为部分要点,请阅读原文,深入理解监管要求。
解读法规指南:ICH M3(R2)《药物非临床安全性研究指导原则》
适用岗位 :
必读岗位:非临床研究部门(Nonclinical Research)、临床前安全评估部门(Preclinical Safety Assessment)、药物开发部门(Drug Development)、注册部门(Regulatory Affairs)。 工作建议:非临床研究部门:确保所有非临床研究遵循M3(R2)指导原则,特别是在剂量选择和研究设计方面。 临床前安全评估部门:基于M3(R2)指导原则评估药物的安全性,为临床试验提供支持。 药物开发部门:在药物开发策略中考虑M3(R2)对非临床研究的要求,确保研究的科学性和伦理性。 注册部门:熟悉M3(R2)指导原则,以便在药品注册过程中准确解释非临床研究数据。 适用范围 :
要点总结 :
非临床安全性研究的国际标准 :强调制定和推广非临床安全性研究的国际标准,以支持药品的人体临床试验和市场授权。3R原则 :鼓励在非临床研究中减少动物使用,遵循减少(Reduce)、精炼(Refine)和替代(Replace)的原则。剂量选择 :明确了在一般毒性研究中选择高剂量的指导原则,包括最大耐受剂量(MTD)和其他适当的限制剂量。探索性临床试验 :提供了对早期探索性临床试验的支持,包括微剂量试验和单一剂量或多剂量试验的设计。特定人群考虑 :对儿科人群、育龄妇女、孕妇等特定人群的临床试验前非临床研究要求进行了特别说明。以上仅为部分要点,请阅读原文,深入理解监管要求。