首页
>
资讯
>
FDA 警告信分析揭示 GLP 合规缺陷和经验教训
出自识林
FDA 警告信分析揭示 GLP 合规缺陷和经验教训
2025-09-17
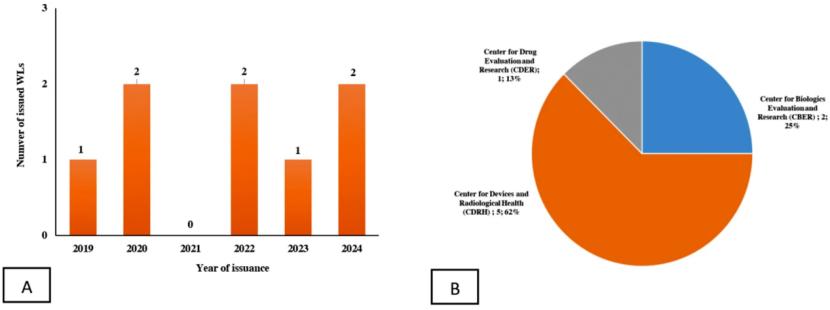
近期,有研究者 在Journal of Pharmaceutical Innovation期刊发表了一项针对GLP合规的FDA警告信的分析研究,文章题为《药物非临床研究质量管理规范在临床前的研究:从2019年至2024年违反21 CFR Part 58发出的警告信中总结的经验教训》 (Good Laboratory Practice in Preclinical Research: Lessons Learned from Warning Letters Issued Between 2019 and 2024 Addressing Violations to the 21 Code of Federal Regulation Part 58),揭示临床前研究实验室在GLP合规方面的常见问题。研究分析了FDA在该期间内由CDER、CBER及医疗器械与放射健康中心(CDRH)发出的8封警告信,涉及临床前研究设施未能遵守21 CFR Part 58 规定的GLP要求。
我国药企往往更关心批准前检查 以及GMP 检查。随着国产创新药走向全球,药企也将面临全世界最高标准的GXP检查。前不久识林介绍了一篇FDA的GCP检查分析文献 ,这次的GLP检查缺陷 分析也特别值得关注。去年我国就有非临床实验室收到警告信 。
尽管只分析8封警告信(大部分是CDRH发出),但缺陷项有67项之多。其中,质量保证部门(QAU)未能履行职责、文件记录不规范、不遵守研究方案、动物处理偏差以及最终研究报告中数据记录问题的发生率均为12%。研究负责人(SD)未能履行职责和不遵守相关标准操作规程(SOP) 的违规发生率各为10%。研究人员资质不足的违规发生率为9%,样品/试剂 及对照品 管理的违规发生率为6%,而与样品/记录留存相关的违规发生率最低,为5%。
违规行为分类与案例
研究负责人职责相关违规(包括未遵守研究方案)
具体案例:某一案例中,SD的职位被当作临时性需求处理,并非是由QAU明确的永久职位。动物健康监测协议要求将动物的疾病、受伤、疼痛和痛苦迹象通知SD和申办方,但未得到执行,部分动物的异常情况未被记录和报告;在另一案例中,协议要求报告并分析体外细胞毒性 测试的所有数据,但实际未涵盖所有数据,细胞活力计算的报告值也不准确。
质量保证部门缺乏适当监督
具体案例:某一案例中,实验室管理层未建立QAU,未通知SD任何偏差 ,也未实施和记录纠正措施。在另一案例中,QAU未明确指定对动物进行麻醉的手术技术人员,未维持必要的潮气量范围,且未审查用于评估动物整体健康的临床病理数据。
人员资质与培训不足
具体案例:某一案例中,FDA检查期间未发现有针对特定方案的培训记录或培训跟踪方法,这使得研究人员的能力受到质疑。在另一案例中,培训文件未能证明研究人员接受了关于记录注射部位不良事件 的培训。
未能遵循标准操作程序
具体案例:某一案例中,检查人员指出实验室未制定关于血液采集和动物护理的SOP,导致测试过程不受控,数据的可靠性受到质疑。
研究动物/样品标识和处理违规
具体案例:某一案例中,研究人员未能准确标识研究动物,且未对动物饮水进行污染物分析,这可能影响毒性测试的结果。
研究记录和样品留存违规
具体案例:某一案例中,FDA检查人员报告称,十二只(12/48)接受被测物质或对照品外科处理的动物相关的“动物麻醉记录”源文件缺失,导致数据无法检索、验证 ,无法重建研究事件。
CAPA措施不能只是培训和改文件
在FDA检查后,场地需对483表 或警告信提出书面回应,并实施纠正与预防措施(CAPA) ,如修订SOP、加强培训或增加人员等。若无法纠正偏差,需说明对数据可靠性 的影响,由FDA评估是否接受。然而,许多CAPA措施被FDA认为不充分,需进一步提供文件或澄清。
以某器械非临床测试场地为例,其违规涉及SD未履行职责,包括未处理呼吸机使用不当导致动物死亡的问题、未遵循方案要求(如未按时检测凝血时间)、未存档研究器械及样本,以及QAU未能有效审查技术操作和数据等问题。尽管场地实施了多项CAPA,如培训、更新文件等,但FDA仍认为其不足,要求补充提供关于异常原因分析、储备样本追溯、人员培训记录 及标签 与存储条件整改的具体证据。此外,该场地在动物标识、水质监测、标本标签与存储条件等方面也存在不合规情形,FDA要求其进一步制定预防策略、评估偏差影响并完善追溯体系。
经验教训总结
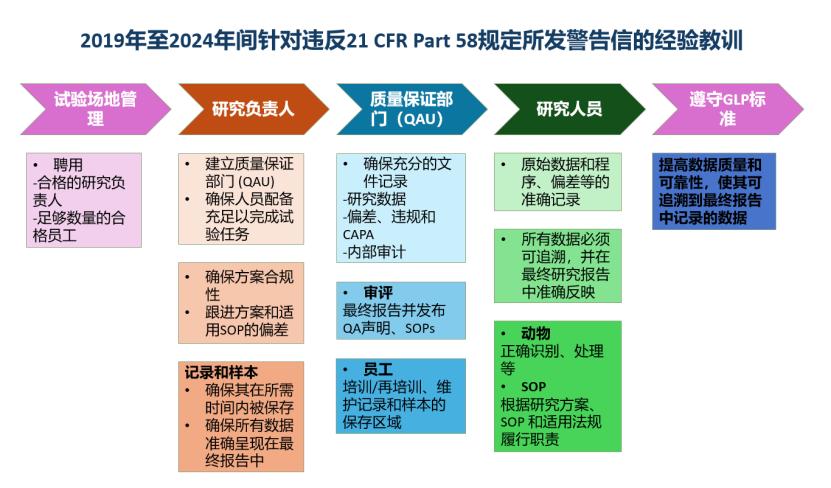
文章最后简要总结了警告信中的经验教训。GLP 的合规始于管理层聘用合格的SD、QAU及经充分培训的人员。其实施依赖于严格遵守研究方案、SOP及21 CFR Part 58 法规。文件质量管理规范(GDP )通过即时准确记录、交叉验证和审计确保数据可靠性与质量。修改记录需划线标注、注明原因并签名日期。专业软件和数据库的应用有助于跟踪记录、样本、培训及审计进程,提升整体合规操作效率。
文章还提供一份经验教训图,先将图片内容翻译如下,供参考
识林-梓
识林® 版权所有,未经许可不得转载
岗位必读建议:
QA(质量保证部门):确保所有实验室操作符合GLP(良好实验室规范)要求。 研发部门:在进行非临床实验室研究时遵守本指南。 临床部门:了解非临床研究的规范,以支持临床研究的顺利进行。 注册部门:在提交研究或市场许可申请时,确保所有非临床实验室研究数据符合GLP要求。 文件适用范围:
要点总结:
GLP规范目的: 确保非临床实验室研究的质量和完整性,以支持产品研究或市场许可申请。适用范围: 适用于所有需要提交给FDA的非临床实验室研究,包括食品添加剂、药品、生物制品等。组织和人员: 规定了实验室人员资质、培训、健康预防措施以及管理层的责任。设施要求: 包括动物护理设施、实验室操作区域和标本数据存储设施的设计和维护。设备维护和校准: 要求对实验设备进行适当的设计、维护、校准和记录。以上仅为部分要点,请阅读原文,深入理解监管要求。