首页
>
资讯
>
FDA 重提创新制造技术战略,当前进展迟缓
出自识林
FDA 重提创新制造技术战略,当前进展迟缓
2025-12-31
12月17日,FDA发布了《FDA创新制造技术的战略文件》 最终版,以履行其在PDUFA VII 中的承诺。该文件基于2023年6月8日由杜克-马戈利斯卫生政策中心主办的公开研讨会意见,并整合了FDA在审评涉及先进制造技术的申报资料中获得的经验。文件更多的是梳理现状,并未提出特别具体的改进举措,不过至少可以看出FDA在业界(通过PDUFA 施加)的压力下,还将继续推动制造技术创新应用。
现有监管机制:各种计划并行,但其实受理和批准并不多
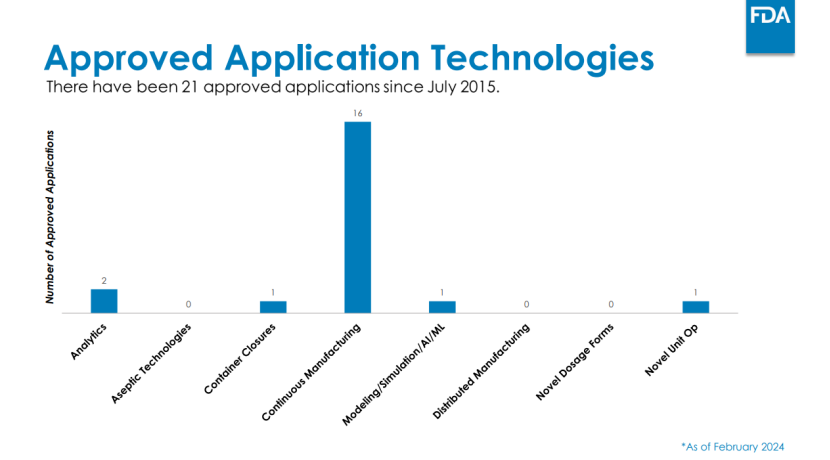
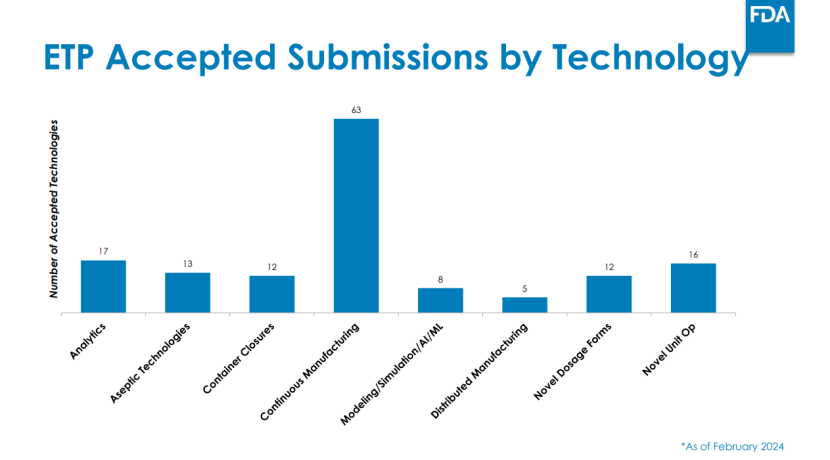
FDA CDER的新兴技术计划(ETP) 及其新兴技术团队(ETT),以及CBER的先进技术团队(CATT),为企业提供了在开发早期与FDA进行非正式交流的途径。ETT通过现场访问、持续技术指导等方式提供支持,其工作已帮助一些企业获得FDA批准,尤其是在连续制造 领域。CATT会议则为CBER监管产品的开发提供了早期建议。
文件中FDA未明确多少企业获批,但在识林收录的2024年2月的一份会议ppt中 ,FDA提供了一些数据可供参考,如下图。
面临的挑战:均不在FDA职权范围内
首先是国际监管标准不统一成为技术全球推广的主要障碍。FDA认为,即便在美国监管要求明确的情况下,生产商对创新技术在海外市场的可接受性仍存疑虑,这直接影响了投资与采纳意愿。
此外,采纳创新制造技术的障碍还来自财务与商业考量。采用新技术需要大量前期投资,而生产商可能资源有限,或对长期投资回报预期不足。这对利润空间较小的仿制药 生产商尤为突出。
行业反馈与建议:核心还是在于降低监管不确定性
FDA注意到行业代表建议FDA加强与国际监管机构的协调,推动形成更一致的监管标准,并通过制度化、公开的对话机制降低全球市场准入障碍。
同时,为激励企业尤其是仿制药企业采纳新技术,行业代表建议FDA针对预竞争领域(pre-competitive spaces,可理解为尚未形成多家竞争局面)的技术经验共享,提供专业法规指南指引,及财政激励措施,以降低不确定性。
对于新设立的“先进制造技术认定计划”(AMTDP) ,业界肯定其以“技术本身”为评估对象的创新模式,并希望FDA在实施中明确数据要求,并对“显著改善”(substantial improvement)等关键概念设定合理预期,以平衡监管的灵活性与确定性。
值得一提的是,截至目前FDA共授予3个先进制造技术认定 ,均为CGT相关技术。
行动计划:可能更新多年前的ETP指南
基于研讨会反馈,FDA制定了三项主要行动计划:
继续加强CDER的ETP和CBER的CATT机制。FDA将更新2017年ETP指南 ,并于2026年底前发布总结报告,同时设立多项绩效指标。CATT已于2024年11月升级为“CATT 2.0”,提升会议处理效率。
识林-梓
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
RA(注册) :必读。确保注册流程符合FDA先进制造技术认定计划的最新要求,及时更新注册资料。QA(质量管理) :必读。根据文件要求,调整质量管理体系,确保生产过程符合先进制造技术标准。研发 :必读。在研发阶段即考虑先进制造技术的应用,以符合FDA的认定计划。适用范围:
文件要点总结:
先进制造技术认定: 明确了FDA对采用先进制造技术药品的认定程序,鼓励企业采用创新技术以提高药品质量和生产效率。注册流程优化: 规定了在先进制造技术认定计划下,药品注册流程的优化措施,以加快审批速度。质量控制要求: 强调了在先进制造技术应用中,对药品质量控制的严格要求,确保药品安全有效。数据完整性与透明度: 特别强调了在制造过程中数据完整性和透明度的重要性,要求企业确保数据的准确和可追溯。监管合作与沟通: 鼓励企业与FDA在先进制造技术应用过程中进行积极的沟通与合作,以促进监管效率。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
CMC(Chemistry, Manufacturing, and Controls)专员 :必读。应深入理解新兴技术在药品制造中的应用,并与FDA的Emerging Technology Team(ETT)沟通,确保CMC信息符合FDA的期望。注册专员 :必读。需熟悉该指南,以便在IND、NDA、ANDA或BLA等监管提交中正确应用新兴技术。研发人员 :必读。应探索新兴技术如何提升产品质量和生产过程,并与ETT合作,确保技术符合监管要求。工作建议:
CMC专员应与ETT合作,解答关于提交信息的问题,并协助进行质量评估。 注册专员在准备监管文件时,应考虑到新兴技术的应用,并与ETT沟通以确保符合FDA的标准。 研发人员应评估新兴技术对产品安全性、身份、强度、质量和纯度的潜在改进,并与ETT合作解决技术或监管上的障碍。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。