当前的许多科学和监管挑战都与非 PK BE 终点的使用有关。例如,使用可比性 PD 或临床终点可能不敏感,无法检测出配方差异或在时间、人力资本和资金投资方面花费巨大。当考虑体外方法时,临床相关的体外检测参数以及评估标准的鉴别会带来巨大的科学挑战。QMM 在帮助仿制药研发商和仿制药审评人员应对这些挑战方面发挥着关键作用。QMM 可以帮助处方和工艺设计、体内 BE 研究的设计以及其它 BE 评价路径的创新。
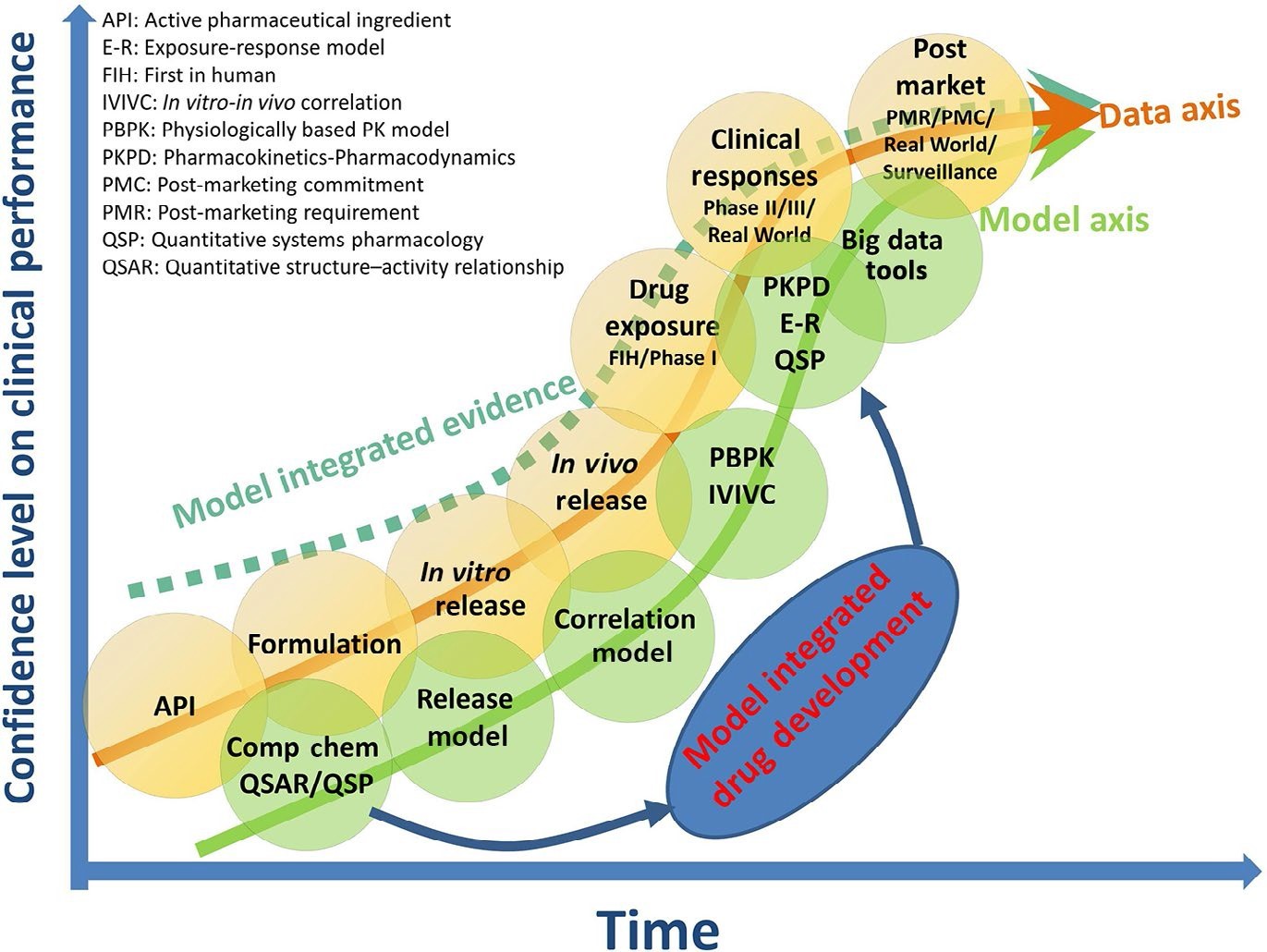
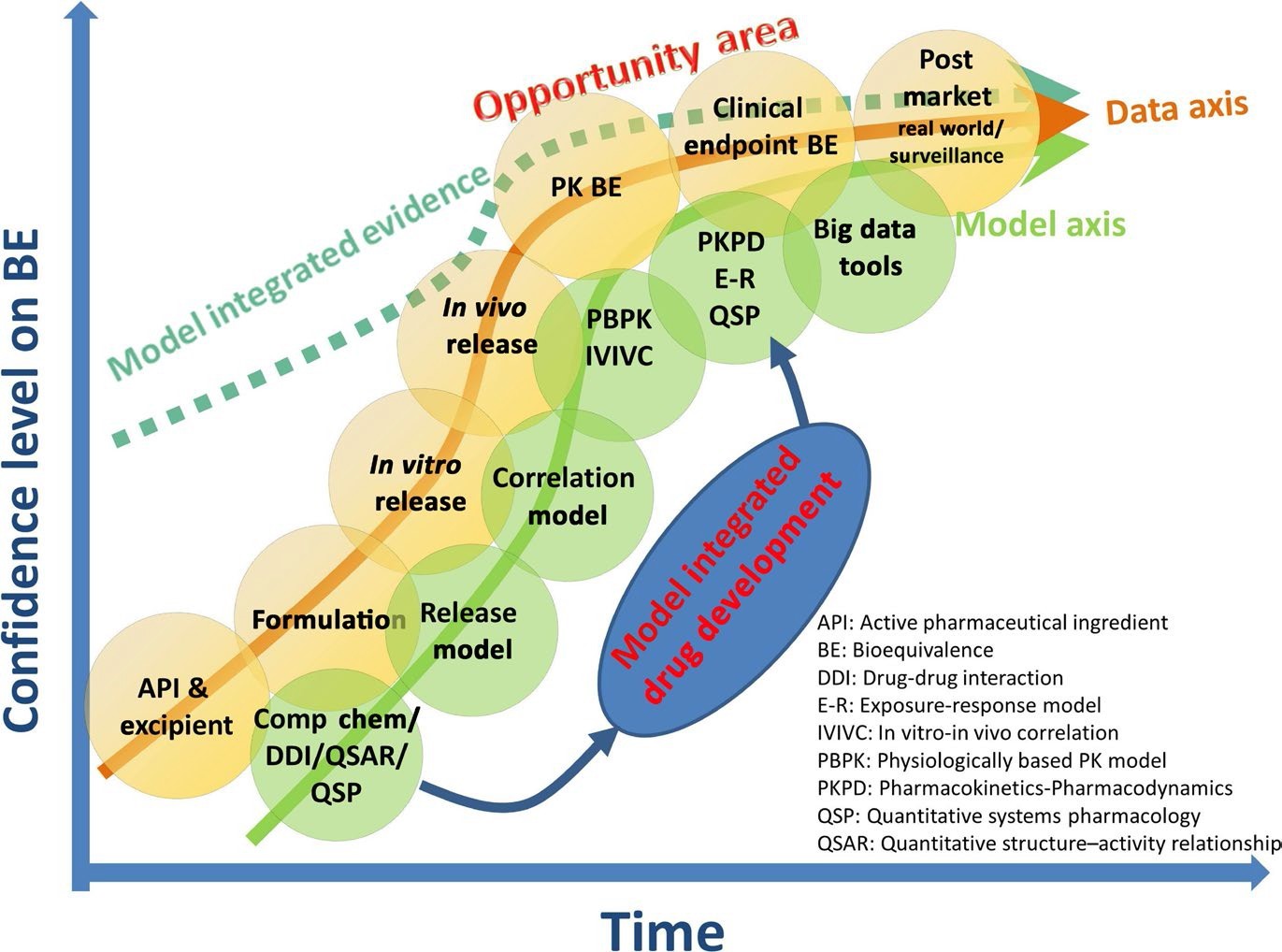
与图 1 所示的新药空间相比,仿制药空间中的 BE 确立(图 2)显示出数据曲线内考虑的三个差异:涉及到新处方时的辅料数据集/信息,PK BE,以及可比性临床终点 BE。与新药研发相似,将收集的数据与建模方法结合使用时,对生物等效性的信心(虚线)可以在更早的阶段达到更高的水平。一个具体的例子是,当建模方法在评价生物等效性时证实了体外数据时。
QMM 包括体外 BE 方法、临床相关质量属性和标准的鉴别,理化(Q3)参数选择,以及体内 BE 标准。所有体内 BE 评价方法均有定量标准,以确保产品在有效性和安全性方面均等效。BE 标准应基于对相应的 PK 变异性和 API 动态 PK-PD/量效反应关系的良好理解。FDA 已经有一些关于 QMM 成功支持了用于窄治疗指数药物4 、高度变异药物5 以及有部分曲线下面积(pAUC)要求的产品6 的示例。
QMM 的下一步是使用模型集成证据为仿制药的批准提供关键信息,或者与相关的体外 BE 评价相结合,作为在适当情况下进行昂贵、耗时且敏感性较低的体内研究的替代方法。与 PBPK、定量临床药理学(QCP)或基于大数据的方法开发相结合,虚拟生物等效性(VBE)模拟已接近成为标准。VBE 模拟的应用范围取决于可用于确认和验证基础模型及其用于预期目的的数据和信息。通过 VBE 模拟,可以生成模型集成证据来协助监管决策。未来 QMM 的应用涉及 FDA 和仿制药行业的利益相关者的参与。FDA 和工业界的科学家应通过在 ANDA 提交前的互动和 ANDA 申报资料包中利用 QMM 来形成现代化仿制药价值链的生态系统,以缩短开发时间并降低成本。所有利益相关者都应充分利用新兴工具(利用大数据分析)来协助产品开发、监管审评和上市后评估。
[1] Journal article “Generating Model Integrated Evidence for Generic Drug Development and Assessment,” by Liang Zhao, Myong-Jin Kim, Lei Zhang and Robert Lionberger, published in Clinical Pharmacology & Therapeutics, Volume 105, Number 2, February 2019.
[4] Jiang, W. et al. A bioequivalence approach for generic narrow therapeutic index drugs: evaluation of the reference-scaled approach and variability comparison criterion. AAPS J. 17, 891–901 (2015).
[6] Lionberger, R.A., Raw, A.S., Kim, S.H., Zhang, X. & Yu, L.X. Use of partial AUC to demonstrate bioequivalence of Zolpidem Tartrate Extended Release formulations. Pharm. Res. 29, 1110–1120 (2012).
Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act)
Mandatory Reading:
Regulatory Affairs (Reg)
Intellectual Property (IP)
Quality Assurance (QA)
Legal Department
Work Suggestions:
Reg: Ensure the company's drug applications comply with the new drug application procedures and bioequivalence standards.
IP: Monitor patent term extensions and the impact on the company's patent strategy.
QA: Verify that manufacturing processes meet the identity, strength, quality, and purity requirements.
Legal Department: Advise on patent infringement issues and the legal implications of abbreviated new drug applications.
Scope of Application: The Drug Price Competition and Patent Term Restoration Act of 1984 applies to chemical drugs, including new molecular entities and generic drugs, in the United States. It is intended for regulatory bodies, pharmaceutical companies, and legal entities involved in drug development and approval processes.
Key Points Summary:
Abbreviated New Drug Applications (ANDAs): The Act allows for the streamlined approval of generic drugs by submitting abbreviated applications showing bioequivalence to the listed drug, without repeating costly and time-consuming clinical trials.
Patent Term Restoration: Offers a mechanism to extend the effective patent life of a drug to partially compensate for the time lost during the regulatory review process, up to a maximum of five years.
Data Exclusivity: Provides a period of data exclusivity, during which the FDA cannot approve ANDAs for other companies that rely on the innovator's safety and efficacy data.
Patent Certification: Requires ANDA applicants to certify about the listed drug's patents or periods of exclusivity, which can trigger a patent infringement lawsuit.
Regulatory Review Period: Defines the regulatory review period for calculating patent term extensions and sets rules for due diligence during the application process.
Conclusion: The Drug Price Competition and Patent Term Restoration Act of 1984 is a landmark legislation that balances the need for accessible, affordable medications with the incentive for innovation. It has significantly impacted the pharmaceutical industry by fostering competition and ensuring that both innovator and generic drug companies have clear pathways to market. The above points are not exhaustive; for comprehensive understanding, the full text of the Act should be consulted.