首页
>
资讯
>
关于阻止Abilify 仿制药上市,FDA给大冢的回复信
出自识林
关于阻止Abilify 仿制药上市,FDA给大冢的回复信
2015-05-11 识林
ANDA引用的参照药品有专利或专营期保护的适应症时(如:3年专营期、7年孤儿药专营期、儿科适应症),ANDA剔除受保护的信息的标签能成功获批吗?
带着这个问题,来看最近正在发生的大冢制药起诉FDA案例,虽然诉讼还没有法院判决,但FDA对大冢的回复信 参照药品 专营期 、专利保护的适应症问题的仿制药同行阅读。
一、背景
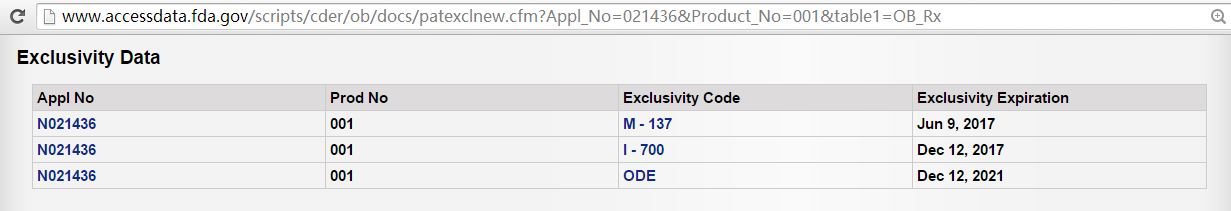
在2014年12月12日,CDER批准了Abilify四个剂型的补充申请,标签上增加了妥瑞氏综合症(Tourette's Disorder)和相关警告、不良反应。此适应症得到了3年专营期(I-700)【橙皮书专营期编码,即:Treatment of Pediatric Patients with Tourette's Disorder(6-18 Years)】
url 和7年的孤儿药专营期(ODE, Orphan Drug Exclusivity)。除了这两个专营期之外,还有一个小儿过敏与自闭症适应症的3年专营期(截至到2017年6月9日)。
2015年4月20日,Abilify的专利将到期。在2015年1月21日,大冢写信给FDA,表明大冢认为FDA不能排除儿科信息,不能在孤儿药专营期结束前批准仿制药。并在2月在马里兰州地区法院起诉了FDA。2015年4月28日FDA批准了Abilify的4个仿制等效产品,分别来自4个公司。FDA也同时公布了对大冢的回复信。
二、FDA总结
三、法律法规背景 a) Hatch-Waxman修正案,列出专利,3年专营期 505(j)(2)(A)(viii) 和21 CFR 314.94(a)(12)(iii) 】
另外,Abilify在最初批准时获得了5年NCE专营期,4年内能够阻止参照Abilify的ANDA递交。此专营期已经结束,不能再阻止ANDA的递交和批准。【参照FD&C Act 505(j)(5)(F)(ii) 】
如果参照药品的补充申请获批,并包括新的临床研究报告,可以获得3年的专营期。这个3年专营期与5年专营期不同。参照药品的5年专营期内不能有ANDA递交,或者4年后,ANDA可以通过专利挑战来递交申请。 3年的专营期不能阻止参照此药品的仿制药递交和获批,事实上,3年专营期只能阻止ANDA包含参照药品通过补充申请获得的变更信息。 ANDA可以在拟定标签中剔除专营期保护的变更,其批准不受3年专营期影响。【参照FD&C Act 505(j)(5)(F)(iv) 】
b) 孤儿药专营期(ODE) 526 和21 CFR 316.20 】。第二步,在药品的罕见疾病适应症获批后,获得7年ODE。
参照FD&C Act 527(a) ,FDA直到7年之后,才能批准此药品此适应症的其它申请。与3年专营期一样是有局限范围的,7年ODE不能阻碍相同药品的未被保护的其它适应症的批准。
c) 获批ANDA产品的相同标签要求 ANDA的标签信息与参照药品已批准的标签相同,除了505(j)(2)(C)请愿批准的不同,或者除了ANDA与参照药品的生产和销售是由不同生产商进行的。
尽管这些条款要求相同标签,但是没有要求同一。相反,这些条款反映出国会的意图是:仿制药标签上的每个处方使用、推荐应是安全有效的,但是不要求ANDA获得参照药品获批的每个适应症。
对于“生产和销售是由不同生产商进行的”的解释,21 CFR 314.92(a)(1) 明确说明:仿制药的拟定使用条件(conditions of use)应与参照药品相同,除了因为专营期或专利而不能批准的使用条件,可以排除这个使用条件。314.94(a)(8)(iv)更明确地举例说明了可允许的标签不同,包括但不限于以下:符合FDA 标签指南或其它指南的有效期、处方、生物利用度、药代动力学、标签修订的不同,或者专利、FD&C Act 505(j)(5)(F) 条款的专营期保护的标签内容。
21 CFR 314.127(a)(7) 进一步说明了,ANDA的标签排除了参照药品被专利、专营期保护的内容,是可以批准的,必须保证拟定药品标签的不同不会比参照药品不安全或不有效 ;即:ANDA不会因为所有保留的、未被保护的使用条件,而不安全不有效。
d) 儿科标签,剔除,免责声明 Pediatric Research Equity Act ),要求已获批成人适应症的,应做儿科人群的该适应症研究。
参照FD&C Act 505B 和21 CFR 201.57(c)(9)(iv)(C) ,如果已批准适用于成人适应症的药,基于充分、良好受控的儿科人群研究而获得了儿科使用的具体声明,这些应在标签的“儿科使用”项下进行总结。
国会授权了FDA,当药品获批了成人适应症(儿童患者也会发生的适应症),但是没有包括充足的该适应症儿科人群使用相关信息,FDA可以判定该药品伪标。【参照FD&C Act 505B(d)(2) 】
FDA担忧的是,已获批成人适应症的药物,也获得了儿科使用的标签,牵涉到FD&C Act 505(j)的因为专利、专营期而进行标签剔除时,应有额外的考虑。当药品获批了适用成人和儿童的同一适应症,儿科信息有专营期保护,并明显与成人使用不同,这时剔除儿科信息、保留成人信息是有安全性风险的。因为在没有充分的安全性和剂量信息下,因为没有依据的期望,可能给儿科患者与成人相同的给药。这种情况下,FDA是不会批准排除儿科信息、保留成人信息的。
为了保证ANDA的批准没有受到延迟(如:参照药品获批用于成人,但相同适应症的儿科使用被Hatch-Waxman法案的专营期保护,且只有儿科使用被保护),国会通过《儿童最佳药品法案》(Best Pharmaceuticals for Children Act )在FD&C Act中增加了505A(o) 。505A(o)当年是为了关闭格华止(Glucophage)可能的专营期漏洞。505A(o) 的标题是“当标签有儿科信息时,推动505(j)药物的批准”,使FDA有法可依;当有Hatch-Waxman专营期保护的儿科标签时,剔除儿科信息可能会有安全性不良后果、并伪标时,应该保证ANDA的标签充分合理,并不被没必要的阻挡。在标签剔除是可行时,这个标签不是限制FDA剔除儿科信息的权力;相反地,是当剔除信息导致不安全,不可行时,它给予FDA权力,可以在ANDA标签上保留Hatch-Waxman专营期保护的儿科信息。
FD&C Act 505A(o)(1)意思是:ANDA申请人排除了505(j)(5)(F)专营期保护的儿科适应症及相关信息时,不应该认为ANDA无资格被批准或者是伪标的。
FD&C Act 505A(o)(2)意思是: 尽管有505(j)(5)(F)专营期条款(关于3年专营期的),505A(o)(1)描述的药物可以获得批准,FDA会要求其声明:(A)该药品的标签不包括儿科使用;(B)为了保证安全使用,包括任何合适的儿科禁忌、警告、预警等信息。
FD&C Act 505A(o)(1) 、505A(o)(2)的条款,表明了国会的意图:剔除3年专营期保护的儿科信息不能确保安全性时,不应该撤销申请未保护适应症的ANDA;即:确保参照药品的儿科标签的保护,不会阻止仿制药进入市场。FD&C Act 505A(o) 的文字意思是:当移除3年专营期保护的信息会导致其它未保护的使用条件不安全和不有效时,会导致伪标时,允许在ANDA标签上保留一些安全性信息。 当移除是可行时,505A(o)不是直接参照的条款。它不是限制,而是补充了FDA长期存在的505(j)标签剔除政策。
编译:识林-榕TM www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
必读岗位:
RA(注册):负责提交儿科研究评估,申请延期或豁免,以及处理标签变更。 QA(质量保证):确保儿科研究的执行和报告符合法规要求。 研发:在药物开发过程中考虑儿科适用性,参与儿科研究的设计和执行。 临床:负责实施儿科临床研究,收集和报告数据。 工作建议:
RA:及时提交儿科研究评估,申请必要的延期或豁免,并处理任何标签变更。 QA:监督儿科研究的执行,确保所有活动符合法规要求。 研发:在药物设计阶段考虑儿科剂量和配方,与RA紧密合作以满足法规要求。 临床:设计和执行儿科临床研究,确保数据的准确性和完整性。 适用范围:
要点总结:
儿科研究要求: 规定了新药和生物制品在提交上市申请时必须附带儿科研究评估,以评估药物在不同儿科亚群中的安全性和有效性。数据外推: 在成人和儿童疾病进程及药物效应足够相似的情况下,可从成人研究中外推儿科疗效。延期和豁免: 在特定情况下,FDA可以推迟或豁免某些儿科研究要求,但需提供合理理由和计划。标签变更: 儿科研究结果必须在药物标签中反映,包括任何豁免信息。信息公开: 所有儿科研究评估、延期、豁免和标签变更信息需公开,以提高透明度。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位 :
注册 :必读。需了解儿科药品市场独占权的延长条件,以及如何通过儿科研究满足监管要求。研发 :必读。在药物研发过程中,应考虑儿科适用性及相应的临床研究要求。临床 :必读。负责儿科临床研究的设计、执行和报告提交。市场 :必读。了解儿科药品的市场独占权对市场策略的影响。工作建议 :
注册 :确保所有儿科研究请求和市场独占权申请符合FDA的最新要求,并及时响应FDA的任何请求。研发 :在药物研发早期就考虑儿科人群的需求,规划包含儿科人群的临床研究。临床 :设计和执行符合FDA要求的儿科临床研究,并确保研究结果按时提交。市场 :利用儿科药品的市场独占权作为市场准入和推广的一部分,同时注意独占权的时间限制。适用范围 :
要点总结 :
儿科研究的市场独占权 :对于新药和已上市药品,如果完成儿科研究并被FDA接受,可以延长市场独占权期限。儿科研究的请求和执行 :FDA可以要求药品企业进行儿科研究,并规定完成研究的时间框架。儿科研究的报告和接受 :企业需在规定时间内对FDA的儿科研究请求作出回应,并在完成后提交研究报告。儿科药品的标签变更 :基于儿科研究结果,FDA可以要求变更药品标签,并有详细的流程规定。儿科信息的传播 :FDA需向公众传播儿科药品研究的信息,包括研究结果和标签变更。以上仅为部分要点,请阅读原文,深入理解监管要求。
Pediatric Research Regulatory Guide Interpretation
Applicable Positions for "Must-Read":
Regulatory Affairs (Regulatory) Quality Assurance (QA) Research and Development (R&D) Clinical Research Work Suggestions for "Must-Read" Positions:
Regulatory: Stay updated with pediatric research regulations and integrate compliance into drug application processes. QA: Ensure that pediatric research conducted by the company meets the regulatory requirements for safety and effectiveness. R&D: Design pediatric studies that are compliant with the regulations and provide meaningful therapeutic insights. Clinical Research: Conduct pediatric studies in adherence to the guidelines and report findings accurately. Scope of the Document:
Key Points Summary:
Pediatric Study Plans: 明确规定了新药和生物制品申请必须附带针对儿科人群的安全性和有效性评估。Therapeutic Benefit: 强调了儿科研究的临床意义,要求研究能够提供对儿科人群有意义的治疗数据。Deferrals and Waivers: 详细说明了在特定条件下,可以推迟或免除儿科研究要求的情况。Labeling Changes: 规定了基于儿科研究结果对药品标签进行变更的流程和要求。Public Disclosure: 要求将儿科研究计划、评估结果和标签变更信息向公众公开,以提高透明度。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读建议:
研发(R&D) :关注罕见病药物的研发策略和申请流程。注册(Regulatory Affairs) :了解罕见病药物的指定流程和条件,确保合规申请。市场(Marketing) :明确罕见病药物的市场定位和潜在优势。文件适用范围:
文件要点总结:
罕见病药物指定请求 :制造商或赞助商可在提交新药申请前请求FDA指定药物用于罕见病或条件。罕见病定义 :影响美国少于200,000人或虽影响超过200,000人但开发成本无法通过销售回收的疾病或条件。生产中断通知 :若药物已获批准,生产中断需提前一年通知FDA;若未获批准但正在进行临床前或临床研究,需通知FDA中断审批流程的决定。公众通知 :FDA需向公众公布药物指定信息。实施细则 :FDA将制定程序以实施罕见病药物的指定流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位
RA(注册):必读。需了解罕见病药物的注册保护政策,以便在注册策略中加以应用。 RD(研发):必读。研发团队需要了解罕见病药物的临床优势标准,以指导药物开发。 QA(质量管理):必读。了解法规要求,确保药品生产和质量控制符合规定。 法律顾问:必读。需要理解法律条款,为企业提供合规建议。 工作建议
RA:在注册罕见病药物时,应充分利用7年的市场独占期,避免在此期间内其他竞争者获得批准。 RD:在药物开发阶段,应着重考虑如何证明新药在疗效、安全性或患者护理方面的临床优势。 QA:确保在独占期内生产的药物符合质量标准,以避免因供应不足导致的批准权丧失。 法律顾问:为管理层提供关于如何利用和保护罕见病药物独占权的法律咨询。 适用范围
要点总结
市场独占权 :对于罕见病药物,FDA可授予7年的市场独占权,期间不批准相同药物的其他申请。例外情况 :在供应不足或持有人同意的情况下,FDA可在7年期内批准其他申请。临床优势条件 :新药必须证明比已批准药物在疗效、安全性或患者护理方面具有显著优势。实施细则 :FDA可制定实施细则,以协助证明临床优势。通知和公布 :FDA需通知药物赞助商关于罕见病药物的指定基础,并在批准后公布临床优势的摘要。以上仅为部分要点,请阅读原文,深入理解监管要求。