|
首页
>
资讯
>
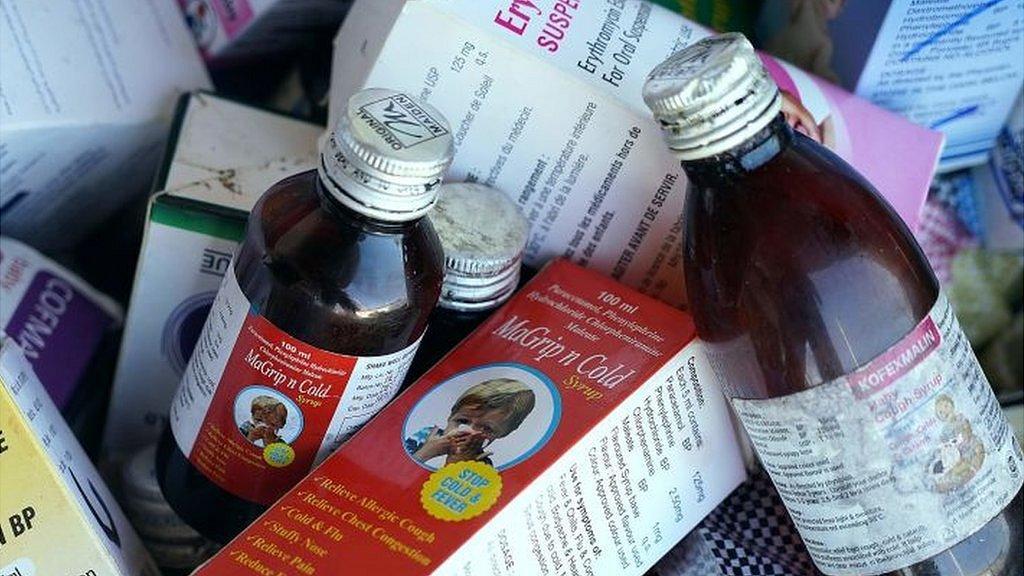
印度药监拟将止咳糖浆移出非处方药目录,应对严重污染风险
出自识林
印度药监拟将止咳糖浆移出非处方药目录,应对严重污染风险
2026-01-08
印度中央药品标准控制组织(CDSCO)于2025年12月29日发布一项修订草案,提议将止咳糖浆从《药品法规(1945)》附表K(Schedule K)中移除。该草案公示期为30天,面向公众征求意见。
根据现行法规,列入附表K的药品类别可在无处方情况下于零售渠道销售。附表K目前列有15类可豁免处方要求的家庭常备药品,其中包含“用于咳嗽的糖浆、含片、丸剂及片剂”。此次修订若生效,将实质终止止咳糖浆在印度部分地区的非处方销售模式。
草案未阐述修订理由,但其决策背景已在2025年11月召开的药物咨询委员会(Drugs Consultative Committee, DCC)会议中说明。会议通报了多起与污染止咳糖浆相关的严重事件,委员会在此背景下审议了附表K修订事项,并批准了移除糖浆豁免资格的建议。
此次监管行动的直接诱因是2025年9月WHO通报的印度境内局部急性疾病聚集事件及多达20名儿童死亡病例。CDSCO随后向WHO确认,在涉事儿童服用的至少三种口服液体制剂中检出二甘醇(DEG)。
印度卫生部门已采取系列风险控制措施。印度卫生部向医师及药师发出指令,要求不得向两岁以下儿童开具处方或调配咳嗽及感冒药品。官方同时指出,原则上不建议对五岁以下儿童使用此类药物。其他相关部门也已行动,防止DEG及另一有毒污染物乙二醇(EG)进入药品供应链。DCC同时审议了制药企业是否应停用丙二醇(propylene glycol)等存在污染风险的溶剂,并要求CDSCO在采取任何监管行动前与相关利益方进行磋商。
DEG和EG污染在人类近现代药品史上屡见不鲜,造成的危害罄竹难书,不仅直接促成了现代药品监管体系的构建,也从未间断地提醒各国药监和业界不可掉以轻心。
以下是对DEG和EG污染和防控相关的重点事件的简要整理,时间由近至远:
- 2025年,印度止咳糖浆污染事件再次引发WHO紧急警报
2025年10月13日,WHO发布警报指出印度生产的部分止咳糖浆遭DEG污染,涉及的药品包括Coldrif、Respifresh TR和ReLife的部分批次。印度已要求涉事药厂停产并召回产品,确认受污染药品未出口,印度中央邦已有至少20名儿童死亡。
由于过去数十年中DEG和EG污染事件频发,EMA于2025年1月更新了EU GMP附录8 原辅包装材料的取样的相关问答。欧盟GMP要求制药企业对原材料进行鉴别检验和质量检查,欧洲药典制定了DEG/EG限度检测方法,企业需对高风险辅料每批检测。制造商需将高风险辅料纳入质量风险管理,了解供应链,除非有科学依据,否则需对每个容器进行检测。新药申请或药品变更时,需确保DEG/EG污染风险得到控制。外用产品如果DEG/EG风险低,可豁免检测。
- 2024-2025年欧洲药典新增检测方法防控糖浆污染
鉴于亚洲和非洲地区持续报告的儿童糖浆受EG与DEG污染致死事件,欧洲药典委员会在2024年3月会议上决定强化监管,于四种常用糖浆辅料(如山梨醇、麦芽糖醇)各论中增设“潜在掺杂”检测章节,规定须使用气相色谱法对EG与DEG进行定量检测,限值分别为620 ppm与0.10%。
继2023年WHO发布警报后,巴基斯坦药品监管局(DRAP)调查发现部分丙二醇存在EG污染,含量从0.76%到100%不等,并于2024年1-3月发布了三次警报,涉及五个受污染批次。DOW公司确认这些物料为假冒,非其生产或供应,质量和安全性无法保证。这些丙二醇被蓄意错标,可能已通过非正式市场流入其他国家,相关制造商可能不知情地购买并储存了这些受污染的原材料。
2023年,USP修订了聚乙二醇(PEG)专论,在鉴别部分新增EG和DEG的限度检测,以防控EG/DEG污染风险。此前PEG专论虽有杂质检测,但不适用于鉴别检验,该修订标准现已正式定稿发布。
2023年5月8日,FDA发布即时生效指南《甘油、丙二醇、麦芽糖醇溶液、氢化淀粉水解物、山梨醇溶液和其它高风险药物成分的二甘醇和乙二醇的检测》,要求对甘油、丙二醇等高风险药物成分中的DEG和EG进行检测。该指南指出,生产商仅依赖供应商的分析报告单难以确保成分安全,强调检测为cGMP合规的必要条件,并建议全面了解供应链及提升员工检测能力。
2022年10月至2023年1月,多国报告儿童非处方止咳糖浆中存在DEG和EG污染,涉及至少七个国家,其中三国报告超过300例死亡,多数为五岁以下儿童。WHO根据各国报告,先后发布了分别针对冈比亚、印度尼西亚和乌兹别克斯坦的全球医疗警报。
此前DEG污染事件
早在1937年,美国就曾发生磺胺酏剂中毒事件,因使用DEG作为溶剂而造成100多人死亡;此后,DEG污染引发的药品安全事件在多国持续出现,1990年尼日利亚、1992年孟加拉、1996年海地、2002年美国、2006年中国(齐二药)和巴拿马均发生过类似事故。
识林-实木
识林®版权所有,未经许可不得转载
岗位必读建议: - QA(质量保证):应全面了解FDA对高风险药物成分的检测要求,确保公司产品符合CGMP规定。
- 生产:需遵循特定身份分析和DEG/EG含量测试的指南,以保证产品质量安全。
- 研发:在药物开发过程中,应考虑原料的安全性,避免使用可能受污染的成分。
- 注册:在药品注册过程中,需确保提交的文件符合FDA对高风险成分的监管要求。
文件适用范围:
本文适用于化学药品和生物制品等高风险药物成分的生产企业,包括原料药、制剂和外包设施,由美国FDA发布,适用于在美国市场运营的大型药企、Biotech公司、跨国药企以及CRO和CDMO等。 文件要点总结: - 合规性警示:强调了制药制造商、复合包装商、再包装商和供应商需警惕甘油和其他高风险药物成分可能受到二甘醇(DEG)或乙二醇(EG)污染的公共健康风险。
- 历史案例回顾:通过历史案例,明确了DEG和EG污染的严重性和检测的必要性。
- 监管要求强调:指出了FD&C法案及其实施条例中关于药物制造的关键规定,特别是与DEG和EG污染检测相关的内容。
- CGMP遵循建议:提供了关于如何遵循CGMP规定,包括对高风险药物成分进行特定身份分析和DEG/EG含量测试的具体建议。
- 供应链管理:推荐制造商维护高风险药物成分的供应链知识,确保成分的来源和分销历史清晰可追溯。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):应熟悉Ph. Eur. 11th Edition中关于质量标准和测试方法的更新,确保产品质量符合欧洲药典的要求。
- R&D(研发):在开发新产品时,需参照药典对原料、制剂和生物制品的指导原则。
- Production(生产):在生产过程中,应遵守药典规定的操作规范和质量控制标准。
适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,包括创新药、仿制药、生物类似药和原料药等注册分类。发布机构为欧洲药典委员会,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 要点总结: - 质量控制更新:Ph. Eur. 11th Edition对多种药品的质量控制标准进行了更新,包括新的测试方法和修订的限值。
- 生物制品指导:新增和修订了生物制品的特定要求,如疫苗和免疫血清的安全性和效力评估。
- 原料药标准:对原料药的纯度、杂质和质量属性进行了明确规定,以确保制剂的安全性和有效性。
- 分析方法验证:强调了对分析方法进行验证的重要性,以确保结果的准确性和重复性。
- 微生物学标准:更新了微生物学测试方法,包括无菌检查、内毒素检测和微生物限度检查。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |