首页
>
资讯
>
国内药政每周导读:ICH 新 GCP 拟实施,化药模块2撰写,疫苗 PACMP,医保形式审查目录,药监数据年报
出自识林
国内药政每周导读:ICH 新 GCP 拟实施,化药模块2撰写,疫苗 PACMP,医保形式审查目录,药监数据年报
2025-09-01
【早期开发与临床】
8.28,【CDE】关于公开征求 ICH《E6(R3):药物临床试验质量管理规范技术指导原则(GCP)》原则及附件1实施建议意见的通知
征求意见截至9月28日。
CDE已定稿中文版E6(R3) ,此次是征求实施建议,即“自 2026 年 3 月 31 日起开始的临床试验,均适用E6(R3) 指导原则。”
更多ICH文件请见“中国 ICH 指南官方翻译和适用公告 ”。
8.28,【CDE】关于发布《疫苗佐剂非临床研究技术指导原则》的通告(2025年第36号)
本文曾于2024年7月征求意见 。
使用“花脸稿”功能可见改动之处较多,建议仔细阅读花脸稿,体会监管导向。
本指导原则适用于感染性疾病的疫苗佐剂,旨在为其非临床研究的设计和开展提供一般性技术建议,以促进新型佐剂以及创新佐剂疫苗的研发。
本指导原则不适用于可对疫苗 其他活性成分起佐剂效应的抗原或通过重组 DNA 技术使之成为抗原分子(如融合蛋白)或免疫原(如载体疫苗)的固有组成部分的免疫刺激分子,也不适用于半抗原或抗原的载体蛋白〔如用于结合多糖的 CRM197、脑膜炎球菌外膜蛋白(OMP)、破伤风类毒素 和白喉类毒素〕。
本指导原则中涉及的新佐剂是指尚未用于已上市疫苗的佐剂,包括以下四种情况:尚未获得安全性信息的佐剂(即全新佐剂)、含有尚未获得安全性信息的全新成分(注释 2)的佐剂/佐剂系统、已获得一定动物和/或人体安全性数据的佐剂、已用于上市疫苗但发生重大变更 的佐剂。
【CMC与仿制药】
8.28,【CDE】关于公开征求《化学仿制药参比制剂目录(第九十七批)》(征求意见稿) 意见的通知
征求意见截至9月10日。
截至目前,参比目录已公示97批 ,发布94批 。识林用户可登录“中国参比制剂库 ”查阅。
【注册与变更】
8.28,【CDE】关于发布《化学药品仿制药上市许可申请模块二药学资料撰写要求(试行)》的通告(2025年第32号)
本文未征求意见,实施日期是2026年3月1日,但CDE“鼓励仿制药申请人 自本通告发布之日起按照本撰写要求提交申报资料。”
文件专门针对化学仿制药 ,包括原料药 和制剂两个附件,指导企业撰写CTD 资料药学部分(即M4Q )的模块二:质量综述部分内容。
化药注册必读。
更多ICH文件请见“中国 ICH 指南官方翻译和适用公告 ”。
另外值得注意的是,2024年4月,CDE曾发布过一套《化学药品3类注册申请药学自评估报告(征求意见稿)》 ,也包括原料药和制剂,其内容与模块二大体相当。据业界专家反馈,这份自评估报告并非申报资料必要部分,目前是看审评人员需求提交。
8.28,【CDE】关于公开征求《预防用生物制品批准后药学变更管理方案技术指导原则(征求意见稿)》意见的通知
征求意见截至9月28日。
本文是为了实施ICH Q12 所发。经查“中国 ICH 指南官方翻译和适用公告 ”页面里的NMPA适用Q12公告 ,24个月的过渡期已在2025年8月到期,Q12正式适用。
本指导原则的主要内容包括批准后变更管理 方案(post approval change management protocols,简称PACMP)的申请、资料要求、拟定变更的研究方案、拟定降低后的变更管理类别、已批准PACMP的修订、按照PACMP实施拟定研究验证工作、不适合提交PACMP 的情形、PACMP 不被批准的情形、PACMP的废止。
上一篇相关指南是发布于2025年6月的《化学药品批准后药学变更管理方案技术指导原则(征求意见稿)》 ,想来治疗用生物制品 PACMP指南也在路上了。
8.28,【CDE】化学仿制药共性问题 - 新增2个问答
2个问题是:
截至2025-08-28,官网共发布化学仿制药共性问题38条(其中有2条为同一共性问题,实际共计37条)。更多相关仿制药问答可查看仿制药质量和疗效一致性评价板块
CDE 常见一般性技术问题解答 、CDE 受理共性问题 也可点击查看。
【市场准入与经营】
8.28,【国家医疗保障局】关于公布通过2025年国家基本医疗保险、生育保险和工伤保险药品目录及商业健康保险创新药品目录调整形式审查的申报药品名单的公告
目录形式审查结果公示期间,共收到75条反馈意见。
根据收集到的意见,我局按程序对有关药品审查结果进行了复核和修正,共有6个药品形式审查结果发生变化,均为申报基本医保目录的药品。具体情况为:
左旋多巴注射液 改为通过形式审查,符合目录外申报条件5;
葡萄糖酸钙氯化钠注射液改为通过形式审查,符合目录外申报条件2;
注射用头孢曲松钠舒巴坦钠改为通过形式审查,符合目录外申报条件2;
熊去氧胆酸口服混悬液通过形式审查,但审查结果改为符合目录外申报条件1/4/5。
最终,2025年目录调整申报阶段医保局共收到基本医保目录申报信息718份,涉及药品通用名633个,535个通过形式审查,其中目录外311个、目录内224个。商保创新药目录申报信息141份,涉及药品通用名141个,121个通过形式审查,其中同时申报基本医保目录和商保创新药目录的有79个。具体药品名单详见附件。
此外,一些非独家品种中,同一通用名不同企业按不同条件申报,依据企业申报条件进行形式审查,只有通过形式审查的药品企业才能参与下一步工作。通过形式审查,仅代表该药品符合相应的申报条件,获得了参加专家评审的资格。只有通过评审、谈判等全部环节的药品,才能最终被纳入目录。
【政策法规综合】
8.22,【NMPA】药品监督管理统计年度数据(2024年)
国家局发布2024年的监管统计年报 ,这里选取一些常见数据,与前4年数据简要对比如下表,供参考(红色字体为最高)。
可以看到:
国产药临床和上市申请不断走高,是未来大批上市的“家底”。
进口化药临床不断走低,上市申请也有大幅下降,与此同时生物制品 不断增多,这也体现了国外新药研发的大方向。
生物制品新药批准创新高,而化药新药批准仅次于2021年。
药企数量还在逐步攀升,监督检查数量有所下降,立案查处数量略有提升。生产环节抽检不合格率略提升。
数据
2020
2021
2022
2023
2024
国产化药临床申请受理
701
1167
1124
1652
1780
国产化药上市申请受理
1012
2473
3099
4887
5940
国产生物制品临床申请受理
369
619
646
908
937
国产生物制品上市申请受理
57
102
88
100
143
进口化药临床申请受理
354
409
361
332
308
进口化药上市申请受理
234
609
470
543
365
进口生物制品临床申请受理
232
204
213
236
256
进口生物制品上市申请受理
66
59
43
95
114
化药创新药临床批准
592
990
1014
1147
1247
化药创新药上市批准
14
24
9
20
23
生物制品创新药临床批准
496
505
571
726
844
生物制品创新药上市批准
2
12
4
15
22
国产文号(化药)
97349
91784
93970
95640
98571
国产文号(生物制品)
1722
1721
1752
1816
1901
药品生产企业(原料药 )
1642
1559
1606
1661
1712
药品生产企业(制剂)
3765
4103
4584
4979
5219
药品生产企业(化药)
3519
3727
4144
4494
4760
日常监督(生产企业)检查家次
19939
17848
18457
18451
16953
完成整改(生产企业)
3262
4043
4590
5262
5193
立案查处(生产企业)
284
227
165
110
160
国家抽检不合格率(化药)
0.43%
0.19%
0.16%
0.40%
0.40%
国家抽检不合格率(生物制品)
0.00%
0.00%
0.00%
0.00%
0.00%
国家抽检不合格率(生产环节)
0.47%
0.31%
0.42%
0.44%
0.62%
以上仅为部分内容,企业可从更多角度比较解读。
【新药批准和报产】
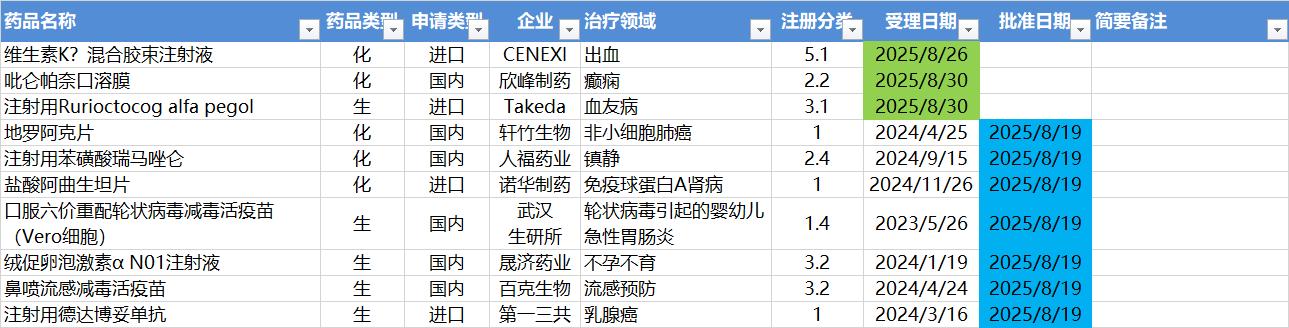
8.25-8.31,NMPA发布7个新药批准,CDE受理3个新药上市申请
注:仅列出新药(包括改良型新药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究 策略、注册路径,都值得业界关注和分析。
识林® 版权所有,未经许可不得转载。
适用岗位 :
QA(质量保证部门):负责监督和确保临床试验的质量管理符合GCP原则。 临床(临床研究部门):负责实施临床试验,确保试验的伦理和科学标准。 注册(药品注册部门):负责向监管机构提交临床试验相关的申请和报告。 研发(研发部门):负责临床试验的设计和试验用药品的开发。 工作建议 :
QA:制定和维护质量管理体系,确保临床试验的每个环节都符合GCP要求。 临床:在实施临床试验时,确保遵循试验方案,保护试验参与者的权益和安全。 注册:及时向监管机构提交必要的申请和报告,确保临床试验的合规性。 研发:在设计临床试验和开发试验用药品时,考虑GCP原则,确保试验设计的科学性和合理性。 适用范围 :
要点总结 :
适用岗位:
注册(RA) :必读。需关注参比制剂目录更新,以便及时调整注册策略。研发(R&D) :必读。需根据参比制剂目录指导仿制药研发工作。质量保证(QA) :必读。需确保仿制药质量符合参比制剂标准。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
以下为对CDE受理共性问题文件的概要分析:
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照更新申报策略,尤其关注多肽类制剂、变更豁免等特殊情形分类规则。 CMC :必读。严格遵循电子申报资料结构(如模块文件夹命名)、签章有效性校验及光盘刻录标准。 临床 :关注临床试验期间剂型变更需重新申报的要求,避免研发中断。 RA :必读。主导跨部门协调,确保电子申报全流程(如申请表签章、资料完整性)符合新规。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照文件调整申报策略,如多肽类制剂按境内外上市情况选择分类,境外变更文件豁免需符合指南条件。 QA :必读。关注非无菌制剂微生物控制标准更新,确保生产工艺与注册标准一致。 研发 :必读。罕见病药物研发需评估是否符合临床试验减免条件,共晶制剂需明确活性成分分类依据。 临床 :必读。临床试验期间剂型变更需重新申请,早期试验适应症填写需备注多队列研究细节。 生产 :必读。批生产记录需包含关键批次,电子申报资料需严格遵循光盘刻录及签章规范。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
临床(Clinical) :必读,负责临床试验的规划、执行和监督,确保合规性。注册(Regulatory Affairs) :必读,负责监管文件的准备和提交,确保符合监管要求。质量保证(QA) :必读,负责监督临床试验的质量管理体系。工作建议:
临床:确保所有临床试验操作符合E6(R3)的最新要求,及时更新SOPs。 注册:在文件准备和提交过程中,特别关注E6(R3)中的变化和新增要求。 质量保证:对临床试验过程中的质量控制点进行审查,确保符合E6(R3)规定。 适用范围:
文件要点总结:
临床试验伦理要求: 明确了临床试验必须遵循的伦理原则,强调保护受试者权益。试验设计和实施: 规定了临床试验设计和实施的具体要求,包括试验方案的制定和执行。数据管理和记录: 强调了临床试验中数据管理和记录的重要性,要求确保数据的完整性和准确性。监查和稽查: 新增了监查和稽查的详细要求,以提高临床试验的质量和合规性。质量管理体系: 鼓励建立和维护一个全面的质量管理体系,以确保临床试验的质量和效率。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证) :应全面理解ICH Q12指南,确保质量体系与监管要求一致,指导产品生命周期管理。注册部门 :需熟悉ICH Q12指南,以便在药品注册过程中有效应用,确保申报材料符合监管要求。研发部门 :应了解ICH Q12指南中关于药品开发阶段的技术和监管考虑,以促进创新和持续改进。生产部门 :需掌握ICH Q12指南,特别是在已建立条件(ECs)和变更管理协议(PACMP)方面的要求。文件适用范围:
文件要点总结:
变更管理框架 :提供了一个框架,以更可预测和高效的方式管理批准后CMC变更。已建立条件(ECs) :明确了MAH与监管机构之间的共识,规定了确保产品质量的要素,以及变更时需要进行监管沟通的条件。变更管理协议(PACMP) :提供了一种监管工具,允许MAH与监管机构就变更所需的信息和监管提交的类型达成预先协议。产品生命周期管理(PLCM)文件 :作为ECs和变更报告类别的中央存储库,捕捉商业阶段产品如何被管理。药品质量体系(PQS)与变更管理 :强调了PQS在管理供应链和产品生命周期中的变更管理的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议 研发(R&D) : 熟悉元素杂质的分类和安全评估原则,确保药物研发阶段符合PDE要求。质量保证(QA) : 监控元素杂质的风险评估过程和控制措施,确保产品质量符合标准。质量控制(QC) : 进行元素杂质的检测和分析,确保检测方法的准确性和可靠性。注册(Regulatory Affairs) : 理解法规要求,准备和提交相关的注册文件,确保产品注册合规。文件适用范围 本文适用于化学药、生物制品、疫苗、中药等药品类型,包括创新药、仿制药、生物类似药、原料药等注册分类。适用于跨国药企、大型药企、Biotech、CRO和CDMO等企业类别。发布机构包括中国、美国、欧盟、印度等。
文件要点总结 元素杂质分类 :根据毒性和在药品中出现的可能性,元素杂质被分为三类,每一类都有特定的考虑因素和控制策略。风险评估 :提出了基于科学知识和原则的风险评估方法,包括识别潜在来源、评估特定元素杂质的存在,以及总结风险评估过程。控制措施 :根据风险评估的结果,提出了控制元素杂质的不同方法,如修改生产工艺步骤、建立规格限制等。分析方法 :强调使用适当的程序来确定元素杂质的水平,确保测试的特异性和准确性。产品生命周期管理 :鼓励在产品生命周期的每个阶段使用基于科学和风险的方法,以持续改进产品质量。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证部门):负责确保文件中提到的质量控制措施得到执行,并监督生产过程中的合规性。 注册(药品注册部门):负责根据文件要求准备和提交药品注册文件。 生产(生产部门):负责按照文件中描述的制造流程和控制标准生产药品。 研发(研发部门):负责药品的药学开发,包括药物的配方开发和生产工艺优化。 工作建议:
QA:检查文件中提到的质量标准和分析程序是否得到遵守,并确保所有生产活动符合规定要求。 注册:根据文件指南准备注册文件,确保所有必要的信息和数据被包含在内。 生产:根据文件中的制造流程和过程控制描述,制定生产计划,并确保关键步骤和中间体得到适当控制。 研发:在文件指导下进行药物开发,包括药物的配方和生产工艺的开发,以及容器封闭系统的评估。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
“注册”:需关注药品目录调整流程和条件,准备申报材料。 “市场”:应根据目录调整结果,调整市场策略和医保准入计划。 “研发”:需根据目录外药品申报条件,规划新药研发方向和进度。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。