首页
>
资讯
>
FDA 公开的已批准药品的283封完全回应函缺陷分析
出自识林
FDA 公开的已批准药品的283封完全回应函缺陷分析
2026-05-06
自宣称将公开完全回应函(CRL )之后,FDA首批试探性公开了一批已批准药品的CRL ,共有202份PDF文档(截至2026年4月30日,已公开了216份PDF文档)。经过对这一数据包的分析,识林提取药品申请编号以及对应的缺陷内容,统计得到202个品种,涉及283封CRL信函,合计产生1,546条缺陷条目。
基于这些缺陷,再从申请类型分布、CRL年份分布、剂型分布、缺陷类型分布等方面进行了统计分析,以期对企业有所帮助。本文仅节选部分内容,完整内容请登录识林查看。
从信函分布来看,平均每个品种收到1.40封CRL,表明相当比例的品种在首次CRL后经历了至少一轮补充审评并再次收到CRL,部分品种甚至经历了多轮CRL交互,反映出企业在回应缺陷时可能存在整改不充分或答复未能完全满足FDA要求的情况。
从缺陷结构来看,1,546条缺陷中批准性缺陷1,141条,占比73.8%;非批准性缺陷(Additional comments)405条,占比26.2%。平均每个品种涉及7.7条缺陷。
统计项目
数量
品种数量(申请编号)
202
缺陷总条目数
1546
批准性缺陷
1141
非批准性缺陷(Additional comments)
405
CRL信函总数
283
平均每品种缺陷条目数
7.7
平均每品种CRL信函
1.4封
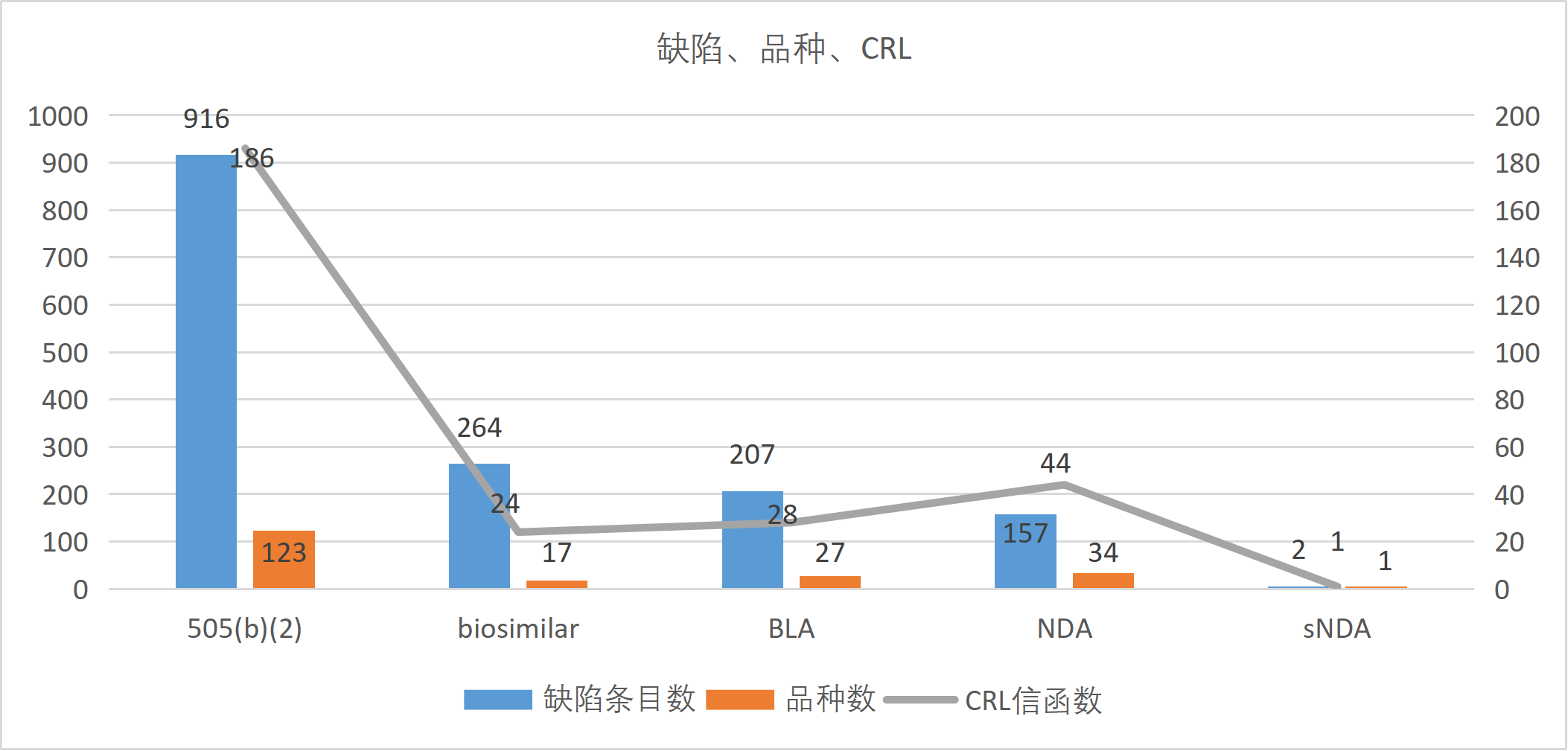
从申请类型看,包括505(b)(2) 、BLA (生物制品许可申请)、NDA (新药申请)、biosimilar (生物类似药)及sNDA(补充新药申请)五种类型。
各申请类型的缺陷条目数、品种数与CRL信函数统计
申请类型
缺陷条目数
占总缺陷比例
品种数
占总品种比例
CRL信函数
占总信函比例
平均每品种缺陷数
平均CRL数量
505(b)(2)
916
59.2%
123
60.9%
186
65.7%
7.5
1.5
biosimilar
264
17.1%
17
8.4%
24
8.5%
15.5
1.4
BLA
207
13.4%
27
13.4%
28
9.9%
7.7
1.0
NDA
157
10.2%
34
16.8%
44
15.5%
4.6
1.3
sNDA
2
0.1%
1
0.5%
1
0.4%
2.0
1.0
合计
1,546
100.0%
202
100.0%
283
100.0%
7.7
1.4
505(b)(2)申请占据缺陷主导地位
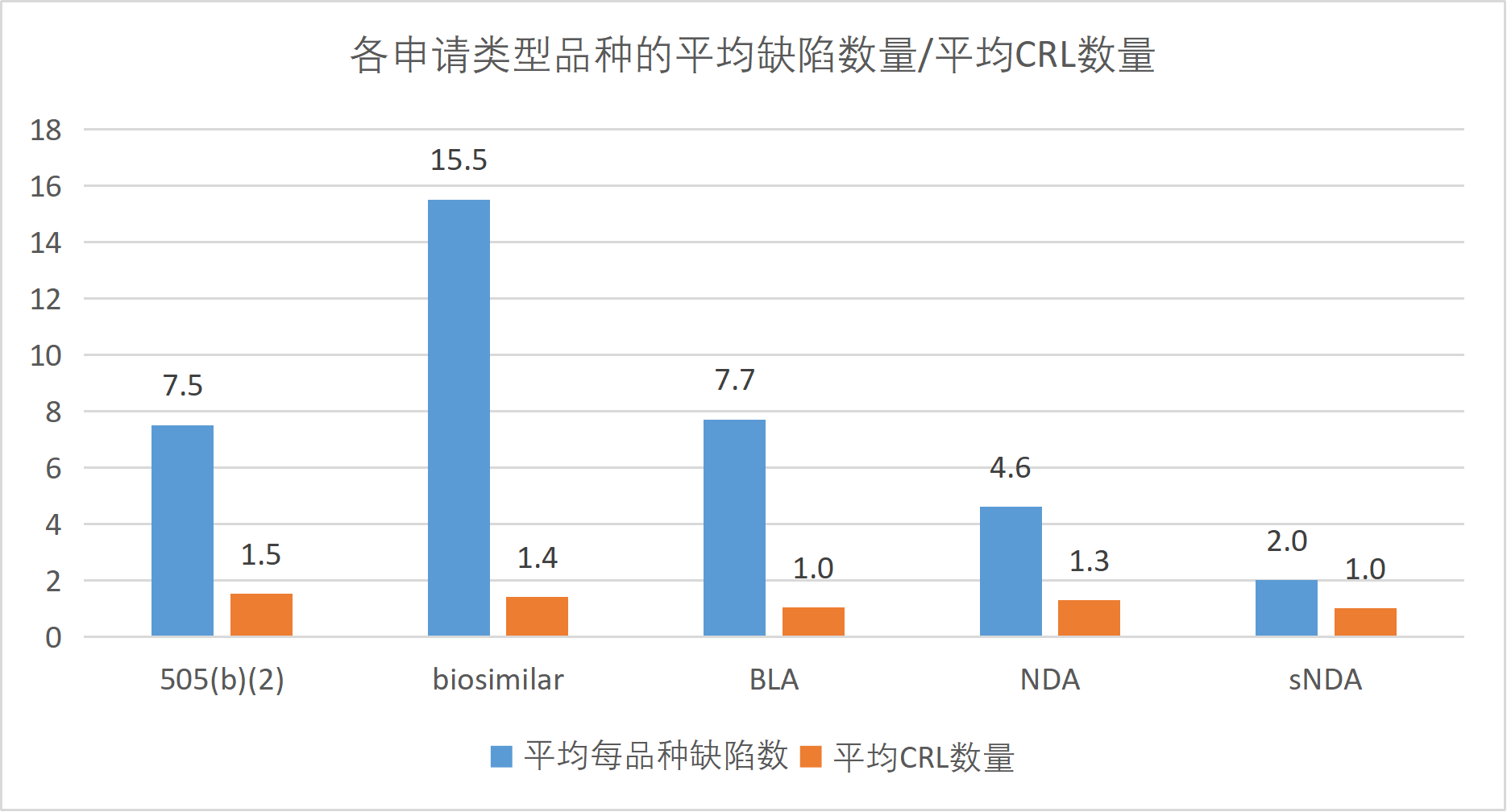
505(b)(2)申请在本数据集中占据绝对主导地位,916条缺陷占总缺陷的59.2%,涉及123个品种(占60.9%)和186封CRL信函(占65.7%)。这一结果反映了505(b)(2) 途径作为FDA审批路径中使用频次较高的类型之一,其固有的监管复杂性。505(b)(2)申请允许申报企业部分依赖FDA已批准药品的安全性有效性数据,但申报企业不拥有原始数据 的"引用权"(Right of Reference),必须通过桥接研究 (bridging studies)证明其改良产品与原研药之间的关联性。FDA对该桥接数据的审查极其严格,企业往往低估了证明改良产品保留原研药安全有效性特征的难度。本研究中505(b)(2)申请平均每品种缺陷数7.5条,高于标准NDA的4.6条,印证了这一审查趋势。
从CRL 信函数来看,186封CRL对应123个品种,平均每品种1.51封CRL,高于总体平均的1.40封,表明相当比例的505(b)(2)品种经历了多轮CRL交互。这提示企业在利用已有数据的同时,仍需投入充分资源确保申报资料的完整性和科学性。
生物类似药单品种缺陷密度最高
生物类似药 (biosimilar)虽然仅涉及17个品种,但缺陷高达264条,平均每品种缺陷数多达15.5条,远超其他所有类型,是标准BLA (7.7条)的2倍。这一显著差异反映了生物类似药审批的特殊挑战。
与小分子仿制药 不同,生物类似药无法通过简单的药学等效性研究证明其与原研生物制品的一致性。FDA要求生物类似药申请提供"总体证据"(totality of the evidence),包括分析表征研究、动物研究及临床研究,以证明其与参照药在安全性、纯度和效力方面无临床意义差异。分析表征中的结构确证(如糖基化 模式、高级结构)、工艺可比性验证以及对免疫原性 风险的评估是FDA审查的重点,也是缺陷集中的领域。
此外,生物类似药的制造工艺复杂,申报企业需证明在无法复制原研药制造工艺的条件下,其产品与参照药 保持了高度相似性。工艺放大 (scale-up)过程中的任何变化都可能影响产品质量属性 ,FDA对此给予格外关注,导致多轮次CRL交互。17个品种对应24封CRL信函(平均1.41封/品种),表明生物类似药的批准路径更加曲折。
不过值得注意的是,生物类似药的“减负”(具体表现为动物试验替代 和临床疗效比对研究 的减免)是当前FDA管理层的核心政策导向之一。这将如何影响具体审评尺度的把握,尚待观察具体案例。
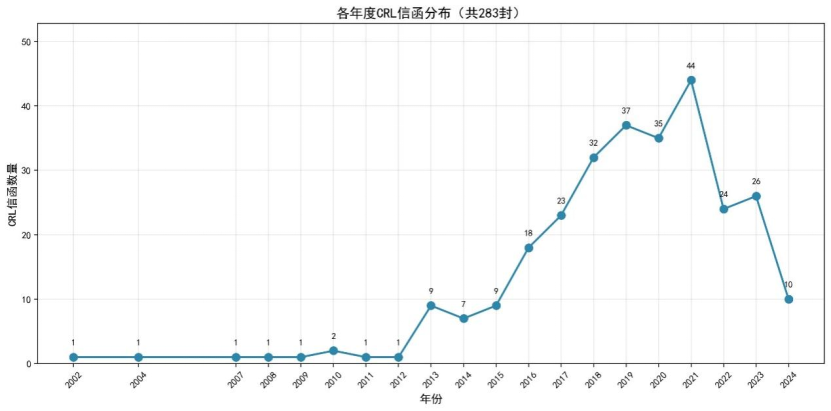
CRL 信函签发年份分布统计
对283封CRL信函按签发年份进行统计,结果如下图所示:
仅针对这批公开的CRL数据,从年度分布趋势来看,CRL信函数量整体呈现先上升后下降的趋势。2002年至2012年期间,每年CRL信函数量极少(不超过2封),这可能与FDA此次对该时期的CRL信息公开程度有限有关(在Drugs@FDA 公开的审评资料中,有更多CRL),也与申报数量相关。自2013年起,CRL信函数量开始显著增长,从2013年的9封上升至2016年的18封。2018年至2021年为CRL信函的高发期,其中2021年达到峰值44封。这一增长趋势可能与以下因素有关:(1)FDA逐步推行CRL信息公开政策,提高CRL信息的可获取性,以近十年的CRL为主;(2)药品申报数量在这一时期显著增加,特别是生物制品 和生物类似药的申报;(3)FDA审评标准日趋严格,导致更多申请收到CRL。2022年至2024年CRL信函数量有所回落,2024年为10封,可能与部分数据尚未完全公开有关,或产品尚未获批(因本数据集为已批准产品的CRL)。
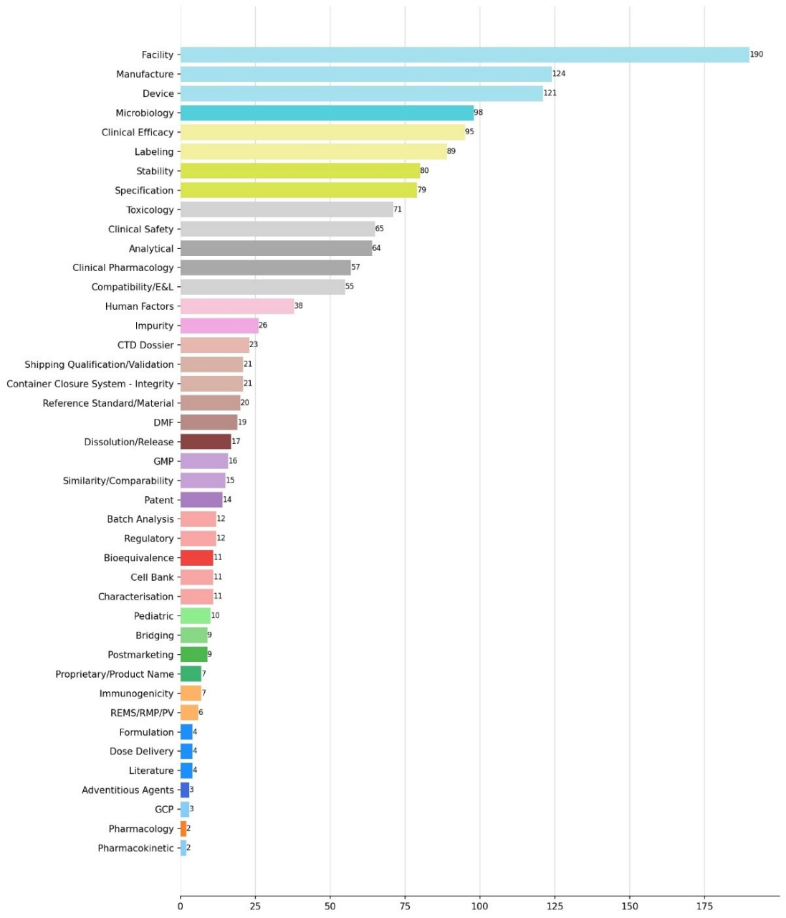
缺陷类型分布统计
将缺陷 内容按照主题进行分类分析,分类药品质量(Product Quality)、设施 检查(Facility Inspections,包含GMP 、GCP 检查)、临床(Clinical)、非临床(Nonclinical)、生物药剂学(Biopharmaceutics)、申请与法律(Application and Legal,包含CTD 资料、专利、监管、产品名称等缺陷)、其它(Others,包含标签缺陷、儿科要求、上市后要求、REMS /RMP/PV要求、文献要求等),统计结果如下表所示:
分类名称
缺陷数量
占比(%)
药品质量
804
52.0%
临床
263
17.0%
设施检查
193
12.5%
其它
118
7.6%
非临床
75
4.9%
申请与法律
56
3.6%
生物药剂学
37
2.4%
总计
1546
100.0%
药品质量(Product Quality)是最主要的缺陷类别,共804条(52.0%),远超其他分类。产品质量问题涵盖生产工艺、分析方法、质量标准 、稳定性 、微生物学、容器密封系统 等广泛的技术领域,是FDA审评中最常见的发补原因。
临床(Clinical)排第二位,263条(17.0%),涉及临床试验设计、疗效数据充分性、安全性评价等。
设施检查(Facility Inspections)排第三位,193条(12.5%),主要涉及生产设施 的 GMP 合规性检查发现问题,以及少量的GCP 检查问题。
需要说明的是,对于缺陷条目的拆分,主要根据 CRL 信函中所列的序号进行拆分,但对于临床方面的缺陷,一条缺陷中经常包含多个问题,并不进行拆分;或因连续多条缺陷相互关联,并且FDA给出解决方案,此时也未进行拆分。因此相对来说,临床缺陷条数和百分比数据存在一定程度低估。
而药品质量缺陷与设施检查缺陷高企。与识林此前的报道 相符。当时针对第二批公开的未批准药物的89份CRL分析发现,有一半新药仅因为CMC 和质量问题遭拒,80%涉及质量缺陷。
此外,进一步的对每一主分类的缺陷内容,做进一步的细分,得到42个子分类。对子分类的缺陷内容的分析整理,请登录识林查看。
作者:识林-樟
责任编辑:识林-木姜子
识林® 版权所有,未经许可不得转载。