首页
>
资讯
>
欧盟延长安全信号检测试点至 2021 年底
出自识林
2020-01-08
安全信号是指新的或已知的可能由药物造成,并需进一步研究的不良事件 的信息,它的来源包括:自发报告、临床研究 、科学文献等。欧盟法规要求上市许可持有人 (MAH)持续监测药物警戒 系统数据,并向欧洲药品管理局(EMA)和成员国药监局报告检测到的经验证的安全信号。
欧盟的安全信号检测试点开始于2018年2月,初始计划时间为1年。2018年10月,EMA和欧盟委员会(EC)第一次延长该试点(为了获得更多经验),但并未给出明确的时间限。2019年12月,EMA和EC同意延长该试点至2021年底。
2010年12月,欧盟通过了新的法令Directive 2010/84/EU和法规Regulation (EU) No 1235/2010,并在2012年6月发布了实施法规Commission Implementing Regulation No 520/2012 (中译 ),提供了操作层面的细节,其中条款18要求,EMA、成员国药监局和上市许可持有人(MAH)应持续监测药物警戒系统数据(Eudravigilance database),并要求MAH向EMA和成员国药监局报告经验证的安全信号。
2017年11月22号,EMA启动了新的药物警戒系统(EudraVigilance),并在2018年2月开始了安全信号检测试点。试点计划要求额外监测(Additional monitoring)药品列表(更新时间:2018/07/18,共293个品种,详细列表请查看EMA topic Signal management)中活性物质 的MAH在药物警戒系统中监测这些药物,并向EMA和成员国药监局报告确认的安全信号。其他未列入表中药品的MAH也可以使用新的药物警戒系统,并从中获取数据,但在试点期间,他们没有持续监测和报告的义务。
EMA应在2019年9月完成试点第一年的报告(该报告还未公开发布),包括工作量和过程。2019年10月的管理委员会会议上,EMA表示,安全信号检测试点主要的结果是,MAH通知了相对较少的安全信号(截止到2018年末为6个,2018全年为114个),并且只有一个值得药物警戒风险评估委员会(PRAC)评估。这些通知给EMA和相关成员国带来了巨大的工作量,可能会将资源从对公共卫生有更高影响的其他活动中转移出去。另外,EMA还补充道,可以通过修订关于新要求的委员会实施法规(Commission Implementing Regulation No 520/2012)来缓和这种事倍功半的情况。下一阶段的计划会包含修改实施法规和延长试点期的讨论,如不把药物警戒信号检测的要求延伸到所有MAH。
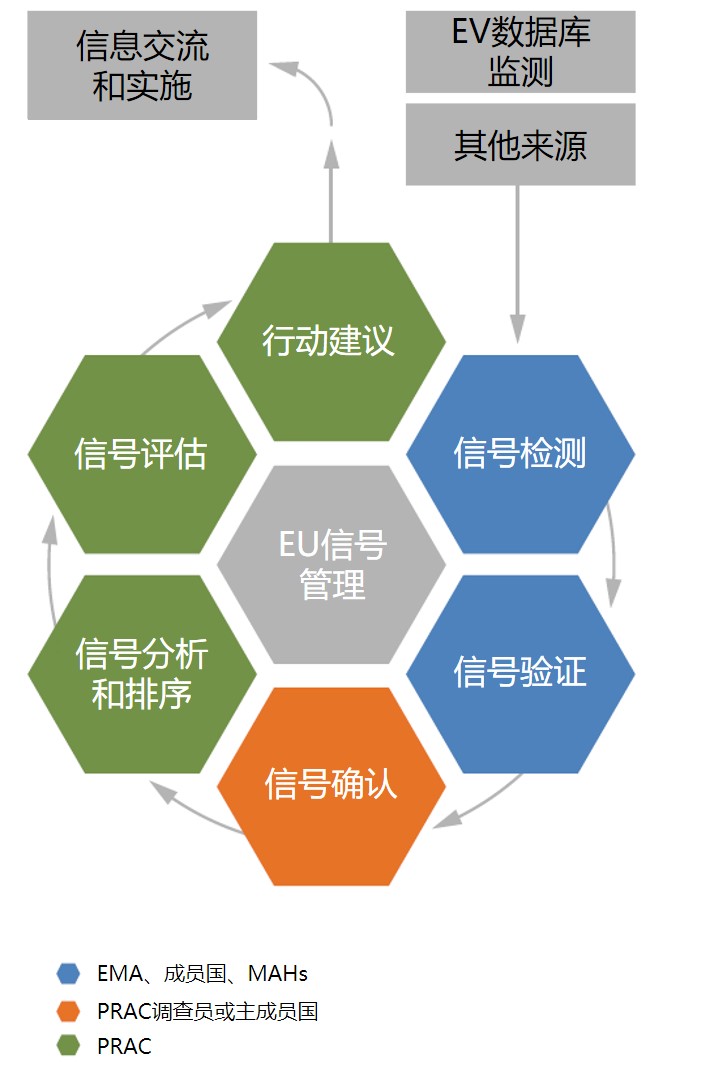
欧盟安全信号管理程序简介
欧盟(安全)信号管理程序(Signal Management Process)主要关注不良结果,基于ICSR的检查、监督系统或研究的整合数据、文献信息等判断是否有与活性物质或药物相关的新的风险产生,或已有的风险是否发生变化。
信号管理包括6部分工作,分别为:信号检测、信号验证、信号确认、信号分析和排序、信号评估和行动建议。其中信号检测和信号确认由EMA、成员国药监局、MAHs共同完成,信号分析和排序、信号评估和行动建议则由PRAC完成。EMA和成员国药监局分别对集权程序获批药物(CAPs)和各成员国内上市药物(NAPs)进行安全信号检测。
潜在的安全信号需要更多数据判断是否需要进一步分析。首先是对所有ICSRs的评估,能获得病人的统计学信息、结果是否致命、是否有其他一同使用的药物、相关的不良反应 等信息。在评估过程中,还需要考虑临床相关性、暴露量、时态关联(temporal association)、生物学似真性(biological plausibility)、停止用药和再激发、不良反应的严重性和结果等。其他的信息来源,如文献和试验结果都可能帮助支持或反驳相关性。
EudraVigilance是潜在安全信号最重要的来源,2018年EMA共审查2204个潜在安全信号,其中78.7%来源于该数据库。数据库中包含的安全信息通过电子反应监测报告(eRMRs)进行持续筛选,其中额外监测药物(即试点中要求MAHs监测和报告的药物)每两周生成一次,其他药品每月生成一次。EMA和成员国每年通常分别要对超过2000个潜在安全信号进行评估,约2%通过验证,如2018年共114个(EMA和成员国分别验证74个和40个)经验证的安全信号。
PRAC对安全信号进行评估后会给出行动建议,包含以下一种或多种组合:
目前不需要进一步评估或行动;
需要更多信息,包括:
监测任何相关新的信息;
对EudraVigilance数据库或其他数据来源进行额外分析;
需要MAH在后续PSUR或递交ad-hoc PSUR,提供更多数据;
由MAH开展上市后安全研究;
需要监管行动,如:
通过变更来更新产品信息(产品特征总结和包装说明书)和/或风险管理 计划;
转介程序(EMA代表欧盟对特定药物或某一类药物类别进行科学评估,该药或该类药物被“转交”给EMA,以便EMA对整个欧盟的提出统一建议);
紧急安全限制;
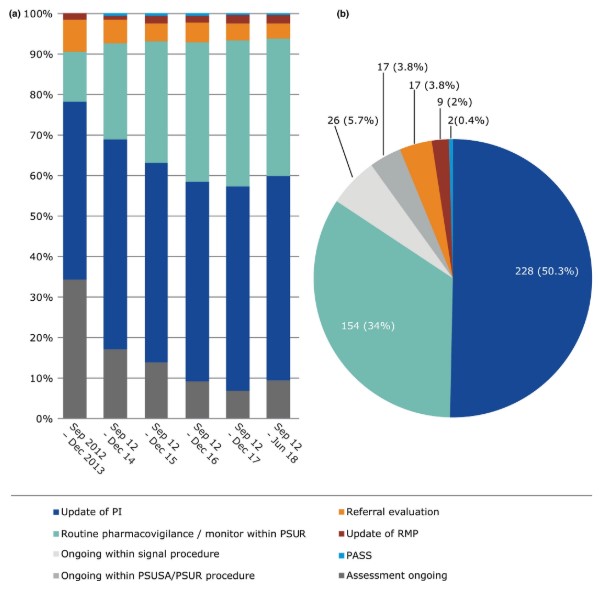
2012.09-2018.06的数据显示,欧盟共处理了超过26,000个潜在安全信号,由PRAC进行评估的共465个,其中50.3%的建议是更新产品信息,34%是PSUR中的常规药物警戒和监测,3.8%为转介程序。
作者:识林-栀
参考资料
[1] Joanne Potts, Georgy Genov, Andrej Segec, June Raine, Sabine Straus, Peter Arlett. Improving the Safety of Medicines in the European Union: From Signals to Action. Clinical Pharmacology & Therapeutics.
[2] Ian Schofield. EU Safety Signal Detection Pilot Extended Again.
[3] EMA topic Signal management
[4] EMA topic Legal framework: Pharmacovigilance
[5] EMA newsletters What’s new in pharmacovigilance - QPPV Update - Issue 2 – 2018.
[6] EMA 2018 annual report on Eudravigilance for the European Parliament, the Council and the Commission.
适用岗位及工作建议:
PV(药物警戒)专员/负责人 :必读。负责确保药物警戒系统主文件(PSMF)的准确性和及时更新,以及药物警戒活动的合规性。QA(质量保证) :必读。监督药物警戒质量体系的建立和执行,确保符合规定要求。注册部门 :必读。了解药物警戒要求,以支持药品注册和监管合规。研发部门 :必读。在药物开发过程中考虑药物警戒要求,特别是在风险管理计划和后期授权安全研究方面。文件适用范围:
文件要点总结:
药物警戒系统主文件(PSMF) :明确了PSMF的结构、内容、维护、文件形式、分包和可用性等要求,以确保药物警戒系统的透明度和可追溯性。质量体系要求 :规定了药物警戒活动的质量体系的最低要求,包括人力资源管理、合规管理、记录管理和审计。Eudravigilance数据库监测 :强调了对Eudravigilance数据库的持续监测要求,包括识别新风险和变化风险、信号管理流程和工作共享。术语、格式和标准使用 :规定了在药物警戒活动中使用国际公认的术语、格式和标准,以促进信息交换和系统互操作性。风险管理计划和定期安全更新报告 :要求制定风险管理计划,并定期更新安全信息,以监控药品的风险-效益平衡。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读建议:
药物警戒专员(PV专员) :详细解读药物警戒系统主文件的结构和内容,确保药物警戒活动的质量体系符合最低要求。质量保证部门(QA) :关注药物警戒活动质量体系的最低要求,确保记录管理与数据保存符合规定。注册部门 :理解Eudravigilance数据库中监测数据的最低要求,以及疑似不良反应报告的传播规定。研发部门 :掌握风险管理计划的内容和格式,以及定期安全性更新报告的要求。文件适用范围:
本文适用于欧盟成员国,涉及人用药品的药物警戒活动。适用于所有药品类型,包括化学药、生物制品、疫苗等,不包括中药。适用于创新药、仿制药、生物类似药、原料药等注册分类。适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。
文件要点总结:
药物警戒系统主文件(PSMF): 强调了PSMF的结构和内容,要求上市许可持有人维护最新状态,并在必要时进行修改。质量体系要求: 明确了上市许可持有人、国家主管机构和欧洲药品管理局应建立的质量体系最低要求,包括人力资源管理、合规管理和记录管理。Eudravigilance数据库监测: 规定了对Eudravigilance数据库中监测数据的最低要求,包括信号管理过程和工作共享。疑似不良反应报告: 规定了个例安全报告的内容和电子沟通格式,要求成员国和上市许可持有人确保报告的完整性和准确性。风险管理计划(RMP): 强调了RMP的内容、摘要、更新和格式要求,以及定期安全性更新报告的规定。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。