|
首页
>
资讯
>
美国FDA和工业界对积压和工作量两术语的纷争
出自识林
2017-03-03
美国FDA仿制药办公室(OGD)和工业界之间关于积压(backlog)和工作量(workload)这两个术语一直有争议,过去几个月中这一争议尤甚。在最近的普享药协会(AAM,过去的仿制药协会)年会上,对这两个术语语义的争执已几乎呈鼎沸之势。那么为什么对OGD在各种情况下描述ANDA数量的术语有那么大的争论,这些情况包括无论是原始未审评的申请还是那些已经由申请人答复的完全回应函,或者是那些等待企业回复的函件?一些争论涉及历史,一些争论涉及感知(optics)。
让我们从历史视角开始。当Hatch-Waxman法案于1984年9月首次通过时,在法案生效前有60天的短暂期限。1984年11月24日,仿制药办公室(当时为仿制药处)可以接收ANDA的第一天,马里兰州Rockville市(OGD所在地)Parklawn楼外的装卸车位上卡车一字排开,超过1000多件ANDA被拖上楼给一小撮专门的联邦雇员等待处置。从那天开始,OGD有180天的时间审评每件申请,我们称之为积压。为什么?不是因为申请已逾期,而是因为这是我们还没有完成的工作。多年来始终使用“积压”术语意味着,ANDA在OGD审评的某个阶段,尚未完成对ANDA的审评,并且尚未向申办人发送审评函、批准或暂时批准函。
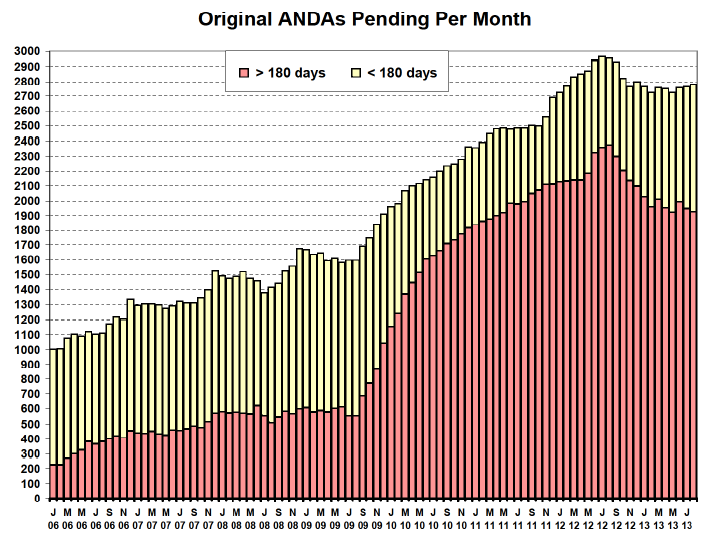
下图是由OGD编制的最后一份“旧格式”的2013年8月月度统计报告(在GDUFA I期改变FDA和行业格局之前的最后一份这种格式的报告),我们可以看到这份报告反映了“每月待决的原始ANDA”的数量,并且包括新ANDA和那些企业已经回应了缺陷函的ANDA。
可以看到,红色部分是超过法定180天审评时间期限的ANDA,黄色部分是还没有敲响180天审评时钟的ANDA。这些ANDA归起来就是我们所称的“积压”。通过在180天审评期内ANDA数量的减少以及一般来说在任何给定月份中未决ANDA总数的减少来衡量进展。
随着GDUFA的通过,情形发生了巨大转变,OGD开始在ANDA审评和批准过程中转型变革。除了审批过程变更外,GDUFA还为“积压申请”提供了一个法定定义,并对于这些在OGD未决的申请或者在2012年GDUFA实施之前企业对缺陷函的回复评估了称为“积压费”的单独收费。但每当FDA和企业谈论“积压”时,事情开始变得有些模糊。一些人继续使用GDUFA下法定意义上的术语“积压”,即在2012年10月1日之前的申请。而企业观察者使用相同的术语来描述等待FDA最终行动的并且已超过其队列年量度日期的所有ANDA。这造成了相当大的混乱,尽管FDA官员已费力澄清其意指的含义。现在随着GDUFA II期的迫近,似乎有更多感知的问题驱动术语“积压”和“工作量”的使用和语境之间的紧张局势。
我们知道,到目前为止FDA已经达到或超过了所有的GDUFA I期目标。但当FDA之外的任何人提及或讨论已提交但未获批的ANDA申请时,这类申请被称之为积压。OGD认为,FDA医药产品使用者付费计划区分等待FDA行动的提交和等待申请人行动的提交。从FDA的角度看,FDA基本上所有ANDA都在GDUFA审评目标或目标行动日期(TAD)内,大多数ANDA已经至少审评过一次。此外,有大量ANDA是在申请人手中延迟而不是在FDA手中。由于申请人对ANDA的优先级排序,许多ANDA在申请人那里待决而可能会延迟返还到FDA。在某些情况下,一些ANDA可能无法继续审评,但这些ANDA却经常被视为积压的一部分,FDA也几乎没法控制或了解这些ANDA对于FDA工作量的真正影响。FDA和企业共同的目标是更快地批准更多仿制药,并且能够达到输入输出平衡的“稳态”。
药品定价问题正在国会热烈讨论,并且成为媒体上的头版新闻。显然,降低药价的一个方法就是批准更多仿制药。因此,每当一个产品的仿制版本获批时,应当并且可能以某种方式有助于稳定或降低特定产品的药价。批准是来自称为“工作量”还是“积压”的申请很重要吗?并不重要。这就是谈论变成感知的地方,在华盛顿,一切事情都与政治感知有关。但是,我们是应该将我们的精力花费在定义在OGD前未决的ANDA上,还是应该只是决定定义一个人人都能理解的单一术语?
也许我们可以这样看。可把GDUFA之前的ANDA看作第0队列年,GDUFA队列年的ANDA就落入到第1-5队列年。如果我们看看对这6个队列采取行动的透明化统计数据,我们可以理解资源消耗在了哪里,以及每个队列年申请随时间推移的进展,我们为什么应该关心这些申请是属于所谓的“积压”还是“工作量”申请?答案显而易见。
那么我们应怎么看呢?我们需要跳过使用任何特定术语,而将讨论集中在第0-5队列年中每年的持续进展和结果。最终目标不是没有待决申请 — 这将意味着企业不再提交新申请,这对于除原研药商外的任何人都无益。为了增加可负担得起的仿制药的供应,仿制药企业绝对必须持续提交新申请。相反,最终目标应是拥有透明的、可预测的审评流程,使得高质量的申请可以在10个月时间框架内得到可靠地审评并且有希望获批。
现在是时候专注于将行业和FDA推向这种理想的稳态了。这需要达成从企业和FDA视角看来都是合理的共识,同时考虑到整个端到端的流程。该流程开始于仿制药公司的研发阶段,贯穿高质量申请的编制和提交,包括FDA对申请的审评,最终获得可以及时进入市场的安全有效药品的批准。这将要求FDA在企业需要及时和一致指南的领域提供明确标准;还将要求企业竭尽全力向FDA审评申请所需的信息;并且将要求双方继续专注于合作解决任何科学、监管或生产问题。我们需要简化和透明。我们需要对进展有信心,需要向FDA提供充足的资源,并确保企业向OGD提供最高质量的申请。为了实现这一目的,双方都需要有所行动,参数需要清晰明确。好消息是,在AAM年会上的讲话中,OGD主任Uhl医师表示了在这些问题上与工业界合作的热情,并强调FDA和企业在达成稳态的目标上协力合作。
为此,AAM及其成员企业坚持将稳健、翔实的绩效报告作为GDUFA II协议的一部分。例如,每月,FDA必须报告输入(提交)与输出(批准和其它行动)。每季度,FDA必须报告等待FDA行动的ANDA数量,以及等待申请人行动的ANDA数量。每年,FDA必须报告平均和中位批准时间,以及财年接收队列中获批ANDA审评轮次的平均数和中位数。请放心,我会像鹰一样观察数据并定期报告有关FDA朝着期望稳态的进展。
原文地址
Lachman CONSULTANTS - Bob Pollock先生
编译:识林-椒
识林®www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
|