|
首页
>
资讯
>
首个 CLDN18.2 靶点药物未获 FDA 首轮批准的原因
出自识林
首个 CLDN18.2 靶点药物未获 FDA 首轮批准的原因
2025-01-09
2024年12月31日,国家药品监督管理局(NMPA)官网显示,安斯泰来的注射用佐妥昔单抗(zolbetuximab)的上市申请已获得批准,是一款同类首创的靶向 CLDN18.2 的 IgG1 抗体。至此,该药已在日本(2024.03)、欧洲(2024.09)、美国(2024.10)和中国(2024.12)获批上市。
犹记得2024年1月,安斯泰来在其官网发布新闻 - FDA 无法在2024年1月12日的处方药使用者费用法案 (PDUFA) 行动日期之前批准 BLA,因为在对 zolbetuximab 的第三方生产设施进行许可前检查后,存在未解决的缺陷。铺天盖地的转发信息,透露出各界人士对于 CLDN18.2 靶点首个药物因 GMP 问题未能获批的唏嘘。
如今,全球主要监管机构都已批准了 zolbetuximab,似乎没有人想再探究当初导致“浪费”了优先审评券的设施问题。
2024年1月未能获批,仅因为设施问题吗?
根据 FDA 公开的审评资料,完全回应函 (CRL) 中描述的导致本轮审评未能批准 zolbetuximab 确实仅因为设施检查问题。此外,FDA 还提出了 3 个小问题,不影响获批,但也建议申请人去做:
- 1. 成品放行的无菌检测方法,描述是每种培养基的检测总体积为10个复溶小瓶,但根据 USP <71>,无菌检测应是每种培养基20个小瓶,每个容器内容物的一半。
- 2. 成品稳定性检测中的容器密封完整性检测,已验证其特异性和检测限,但 CCIT 方法不是药典方法,还应验证其精密度(重复性、中间精密度和重现性)及稳健性(方法不受方法参数故意变化影响的能力)。需为拟议的染料渗透方法提供完整的 CCIT 方法验证结果。
- 3. IMAB362 成品的商业化生产计划 (b)(4)评估了 (b)(4)。请提供额外的数据和/或信息以支持 (b)(4) 是可以保证 IMAB362 成品生产的重现性的。
那么如何获得设施问题的详情?
要了解批准前检查中的具体缺陷,需要找到 FDA 签发给这家第三方场地的 483,那就需要知道场地名称、检查时间。
① 从 FDA 的审评资料中,可以在批准信中查到 zolbetuximab 的几个时间点,那么检查日期就在2023.05.12 - 2024.01.04 时间段内;
| 首次递交
| 2023.05.12
|
| CRL
| 2024.01.04
|
| 重新递交
| 2024.05.09
|
② 在产品质量审评文件中,查到 DS 和 DP 分属两个场地,但具体场地信息被涂黑了,未能获得;
这时可以查询 EMA 审评资料,看看是否能获得被遮盖的信息。在 EMA 的评估报告中描述:
- 2024 年 6 月,在用于 DS 生产活动的场地进行了一次批准前 GMP 检查,检查结果于 2024 年 7 月 22 日公布。Zolbetuximab 的 DS 由 Patheon Biologics LLC(地址:4766 LaGuardia Drive, Saint Louis, Missouri 63134-3116 United States,Patheon)生产。在评估期间,提出了一项重大异议 (MO),要求通过现场 GMP 重新检查确认 Patheon 场地符合 GMP 要求,可执行用于 Vyloy 的生产活动。进行了 GMP 检查,并收到确认,该场地可被视为符合 GMP 要求,MO 被认为已解决。
由此可推测,大概率是上述 DS 场地导致 FDA CRL。
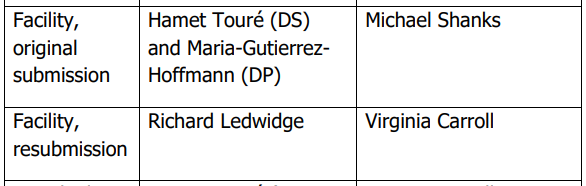
③在产品质量审评文件 BLA Executive Summary - OPQ Review Team 中,查询到,在原始审评阶段、重新提交阶段批准前检查的检查员的姓名。并由此推测,比较可能的情况是,在原始审评阶段仅 DS 场地出现重大问题,在重新提交阶段,对 DS 场地进行了再次检查。
以上3条信息,只要能确认其中的2条,就可以通过识林483数据库,筛选出相应的 483 文件;或者在①②条信息确认的情况下,通过 FOIA 途径向 FDA 索要 483 文件。以下截图,可以确认这封 483 是导致 zolbetuximab 未能在 FDA 首轮审评中获得批准的原因。
导致 FIC 未获批准的 GMP 缺陷
这封 483 包含六大缺陷项,十九条小项。检查时间为 2023/11/03, 2023/11/06 - 2023/11/10
1. 标准操作规程和方案不充分和/或未被遵守
a. DS 上游制造工艺在 2 号楼的 (b)(4) 车间进行,该车间是一个多产品和多工艺设施。目前,(b)(4) 批次的 (b)(4) 不同产品,包括 (b)(4) 的 (b)(4) 细胞培养工艺在 (b)(4) 车间进行。当新产品引入车间时,产品之间的交叉污染风险评估和变更控制未得到充分执行。未遵循 2023 年 4 月 5 日生效的 SOP 000016298 版本 29.0 的变更控制概述。
b. 生产 (b)(4) 的工艺性能确认(PPQ)方案 (Doc No. STL-VAL-PV-0030) 描述了 (b)(4) 和 (b)(4) 生物反应器应在每次运行时更换,以确保其在 (b)(4) 工艺中可接受使用。在执行 PPQ 时,2 号楼 (b)(4) 车间共有 (b)(4) 和 (b)(4) 生物反应器。在 USP PPQ 报告中,表2,只有 (b)(4) 生物反应器经过了验证。PPQ 研究的执行并未遵循 PPQ 方案。
c. 总数为(b)(4)的生产生物反应器用于生产商业批次。但用于生产商业批次的(b)(4)生物反应器,没有包含在工艺验证/确认活动中。DS 的生产工艺验证不充分。
d. SOP-000015807,预防性维护计划,版本 8.0,于 2023 年 7 月 26 日生效,允许对于维护频率为 (b)(4) 的设备,在完成预防性维护到期日之后安排 (b)(4) 次。
e. 2023年11月9日,在 1 号楼 1-122 房间的 (b)(4) 日志,发现主管人员/指定人员未对已完成的页面进行审核。此偏差已添加到 2023 年 11 月 3 日开启的现有偏差 QR-676045 中((b)(4) HPLC-(b)(4)柱日志,QCA (b)(4) 日志,QC温度计使用维护日志,(b)(4)天平性能检查页未能在(b)(4)天内及时审核)。您未能遵守 2023 年 9 月 20 日生效的 SOP-000016285 版本 122。此外,根据 2023 年 6 月 26 日生效的合规巡检和流程确认程序 SOP-000283834 版本 2,质量控制实验室的合规巡检区域关键性很高。应对高风险区域进行 (b)(4) 合规性检查。缺少主管审核的 pH 计日志页记录于 2023 年 4 月。主管审核于(b)(4)已逾期。(b)(4) 合规巡检不充分,因为它未发现错过的主管审核。
f. SOP-000015841 确定要从物流管理中移除的材料 115.0 版(于 2023 年 9 月 22 日生效)并不充分。SOP 规定了在到期(或材料状态变为“拒绝”)(b)(4) 天内启动报废的时间表,如果材料在到期(或接受“拒绝”状态)后 (b)(4) 天内仍在库存中,则将拒绝状态上报给工厂领导团队。但是,(b)(4) (材料 (b)(4),到期 (b)(4),报废表格于 2023 年 1 月 29 日启动) 和 (b)(4) (材料 (b)(4),到期 (b)(4) 和 2022 年 12 月 17 日,报废表格于 2022 年 1 月 29 日启动)直到 2023 年 11 月 6 日当周才获准处置。
g. SOP-000015782,害虫控制政策,版本 115.0,于 2023 年 3 月 22 日生效,不充分。仅当在三周(滚动)时间内在同一区域(即连通房或相邻房间)发现三次昆虫,在六个月(滚动)时间内在同一区域发现六次昆虫,或在一年(滚动)时间内在同一区域发现三次两栖动物、爬行动物、鸟舍或啮齿动物时,才会在生产区域启动偏差。因此,在 2 号楼控制区域中的以下害虫昆虫回收没有启动偏差:
| 2021 , Q3:
| 4
|
| 2021 , Q4:
| 3
|
| 2022, Q1:
| 1
|
| 2022, Q2:
| 12
|
| 2022, Q3:
| 3
|
| 2022, Q4:
| 7
|
| 2023, Q1:
| 2
|
此外,2021 年第三季度在 2 号楼 1700 走廊发现啮齿动物时,并没有启动偏差。1700 走廊不受控,但通过气闸 (B2-1826 和 B2-1821) 与制造套件相连。
h. 检查期间,发现有人员在(b)(4)接触时间结束前或不知道(b)(4)接触时间是否结束的情况下,走在 2 号楼生产区域已清洁过的地板上。
2. 质量保证对 CAPA 有效性的监督不充分
a. 自 2020 年 1 月以来,2 号楼生产车间的 (b)(4) 模具板回收中发生了 27 次模具偏差,导致 27 次偏差及其 CAPA。到目前为止,针对这 27 次偏差采取的纠正措施均未改变以下趋势结果。
| 2020:
| (b)(4) 板回收,7次偏差
|
| 2021:
| (b)(4) 板回收,1次偏差
|
| 2022:
| (b)(4) 板回收,8次偏差
|
| 2023 至今:
| (b)(4) 板回收,11次偏差
|
b. 自 2020 年 2 月以来,2 号楼生产车间天花板已发生 36 次漏水,导致 36 次偏差及其 CAPA。迄今为止,针对 36 次偏差采取的纠正措施未改变以下趋势结果。
| 2020:
| 生产车间天花板漏水 8 次
|
| 2021:
| 生产车间天花板漏水 9 次
|
| 2022:
| 生产车间天花板漏水 8 次
|
| 2023 至今:
| 生产车间天花板漏水 11 次
|
c. SOP-000383867, 版本1.0,于 2023 年 6 月 30 日生效,(b)(4) ,适用于所有在场地的色谱和(b)(4)分析,这份 SOP 是由 CAPA 533317 而来,其已于2023年5月31日关闭。然而,截至目前,(b)(4)峰的相关内容尚未在以下附属的整合 SOP 中实施:
- SOP MET-000005641,版本3.0,于2023年8月23日生效;
- SOP MET-000005648,版本4.0,于2023年4月26日生效;
- SOP MET-000005729,版本6.0,于2023年8月2日生效。
此外,在以下检测方法中,仍未实施以下指导内容:
3. cGMP 物料的标签不充分
a. 2023年11月3日,发现B1仓库的(b)(4)材料的两个袋子上没有标签。2023年11月6日,EHS 人员识别了材料是(b)(4),并处理了仓库里的材料。
b. 2023年11月7日,(b)(4) 批次的物料的一个袋子(物料(b)(4))储存于5车间室温下。(b)(4) 的标签没有储存条件。
c. 2023年11月6日,装有树脂 (b)(4)(材料(b)(4))的 (b)(4) 柱,储存于(b)(4)车间的室温下。柱的日志和柱标签显示储存条件为(b)(4)℃。室温下的储存时间没有得到适当控制。
4. 设备维护不当
a. 2023 年 11 月 6 日,观察到套件 (b)(4) 中色谱仪 (b)(4)(序列号 (b)(4))的最近预防性维护 (PM) 日期为 2022 年 9 月 29 日;该设备的下一个 PM 到期日期为 2023 年 9 月 30 日。该设备的当前供应商 PM 间隔为 (b)(4),包括 (b)(4) 和分析仪器校准。对于该设备,到期日后完成 PM 的额外时间为 (b)(4)。即使包括允许的额外时间,该设备的 PM 也已过期。
5. 在拟议的强制条件下的 zolbetuximab DS 稳定性检测研究未正确进行
a. 2023年11月10日,对用于 (b)(4) DS 拟议长期(b)(4)条件和(b)(4)条件稳定性研究的设施和设备进行了现场检查。对于(b)(4)的(b)(4)条件下的研究,在 1 号楼 1-144 室公共区域的稳定性室(b)(4)中进行的相对湿度 (RH) 检测发现,拟议的(b)(4)DS 稳定性检测容器中的稳定性样品保存在 (b)(4) 中。(b)(4) 密封良好。在观察到的条件下,无法充分达到或维持拟议的 (b)(4) RH;因此,这表明稳定性研究未在拟议的 (b)(4) 条件下进行。此外,(b)(4) 是在 2019 年启动稳定性研究后于 2021 年实施的,缺乏充分的评估或变更控制。两者对拟议强制条件下的稳定性研究产生了负面影响。
6. 对培训的质量监督不充分
a. 对缓冲液/培养基小组一名新员工的 (b)(4) 培训记录进行审核时发现,培训计划与系统记录中反映的到期日期不一致。
b. 对缓冲液/培养基小组三名员工的(b)(4)培训记录的审核显示,2021年、2022年和2023年的培训均在规定的到期日之后完成,或者2023年的培训目前已逾期。
c. 2023 年 6 月 30 日生效的 SOP-000383867 (b)(4) 1.0 版培训未及时纳入培训课程。
总结
在 FDA 签发的此封 CRL 中,未提出任何有关临床或非临床方面的缺陷问题,这对于新靶点的首个新药来说很不容易,也意味着前期的研发所付出的巨大努力。但同时,无论一个创新药多么令人“振奋”,生产和质量管理的短板都可能成为获批道路上的致命障碍。特别是在涉及多方合作或委托生产的情况下,企业必须加强对第三方生产设施的审计和监督,确保其持续合规。只有将 GMP 合规性和生产质量视为药物开发的核心战略部分,才能避免类似的延误或失败。
识林-樟
识林®版权所有,未经许可不得转载。
岗位必读建议: - QA:负责确保实验室操作符合质量控制要求,监督取样、留样、检验等流程。
- 研发:在设计质量标准和分析方法时,需遵循本文规定。
- 生产:在取样和留样过程中,应遵守本文的详细规定以保证产品质量。
文件适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,包括原料药、中间产品、待包装产品和成品。适用于创新药、仿制药、生物类似药等注册分类。适用于中国药企,包括Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 实验室职责与布局:明确了质量控制实验室的职责、布局原则和要求,以及人员的组织架构和资质要求。
- 取样与留样管理:规定了取样过程的控制和留样的定义、量、储存要求及记录。
- 物料和产品检验:强调了检验要求,包括待检样品核对、检验、记录和报告书的编制。
- 委托检验管理:阐述了委托检验的原则、应用范围、职责和工作流程。
- 质量标准建立:详细说明了质量标准的设计与制定、审核与批准流程。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |