首页
>
资讯
>
资讯公开
>
2015年美国FDA新药审批一览
出自识林
2015-12-25
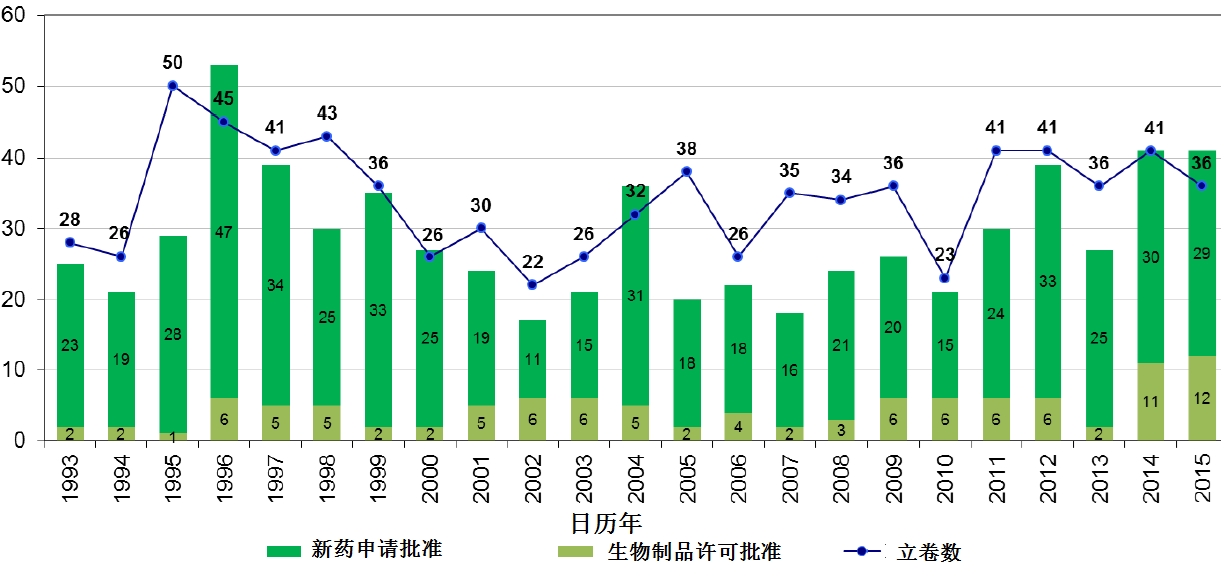
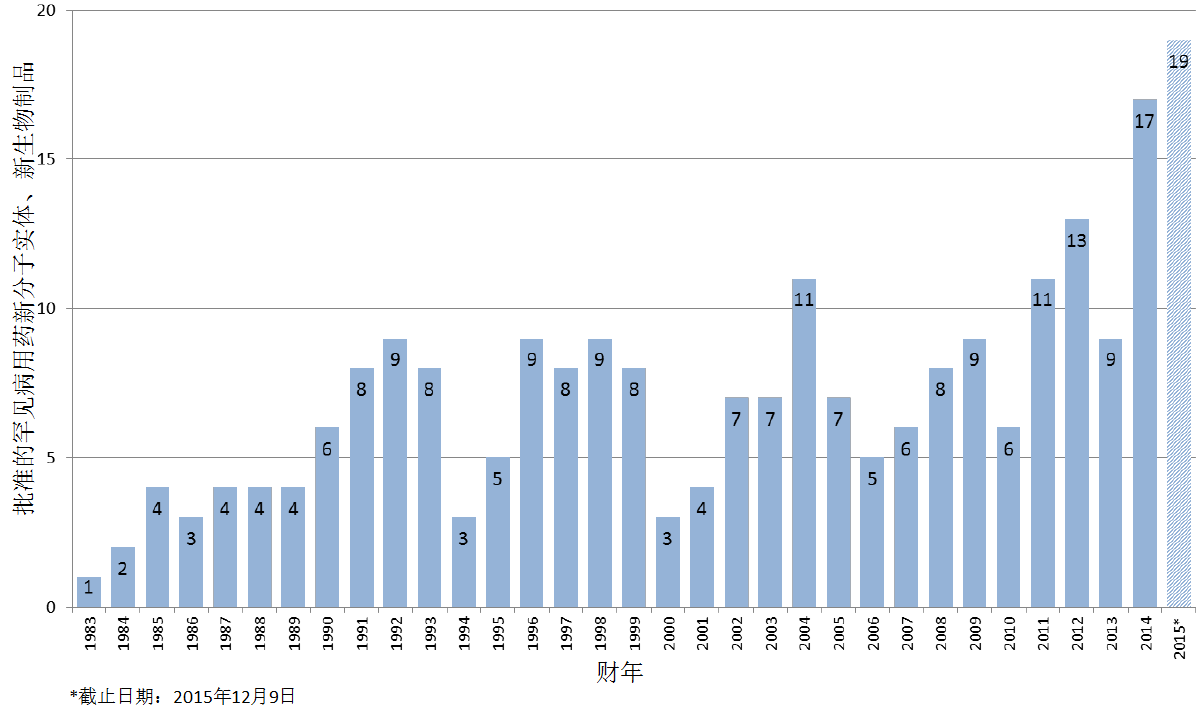
编者按:截止2015年12月9日,2015年美国FDA药品审评与研究中心(CDER)受理36份新分子实体申请;批准41个新分子实体,其中包括19个罕见病用药 。首轮获批率创历史新高。完成PDUFA V规定的新药绩效指标。本文以图文并茂的方式,对2015年CDER新药审批情况作全景式的展现。
2015年12月14日,FDA CDER新药办公室主任J. K. Jenkins博士在2015 CMS/FDA峰会上报告2015年CDER新药审批的最新情况。Jenkins博士从CDER如何实现PDUFA绩效目标、新药审批趋势(IND 、新分子实体申请提交、新分子实体批准;加快审评 计划的行动及效果),以及PDUFA V/FDA安全与创新法案计划所涵盖的新分子实体审评计划和突破性治疗药物认定计划的角度介绍了2015年CDER新药审评情况。
2015年CDER新药审批呈现以下特点:
根据2015年3月Eastern Research Group公布的《PDUFA V计划评价中期报告》,PDUFA V中的新分子实体计划被认为获得广泛成功。突破性治疗药物认定/批准持续增长。2015年4月,布鲁金斯学会开会研讨突破性治疗药物认定,总结实施该审评通道两年半以来的得失。优先审评券 受到持续关注。新分子实体首轮批准率创历史新高。生物类似物继续呈现上升势头。
尽管成绩斐然,但面临的重大挑战依然存在。主要体现在,不断增加的工作量使得计划资源紧张;招聘和留住人才也仍然是所面临的重大挑战。
一.新药审批情况:
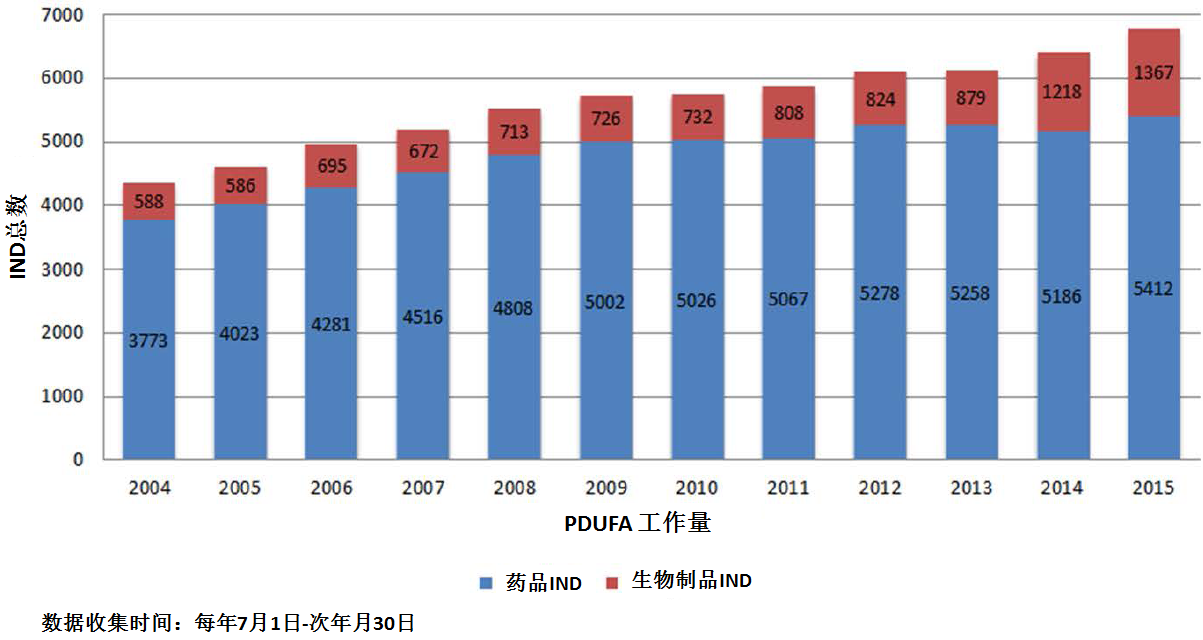
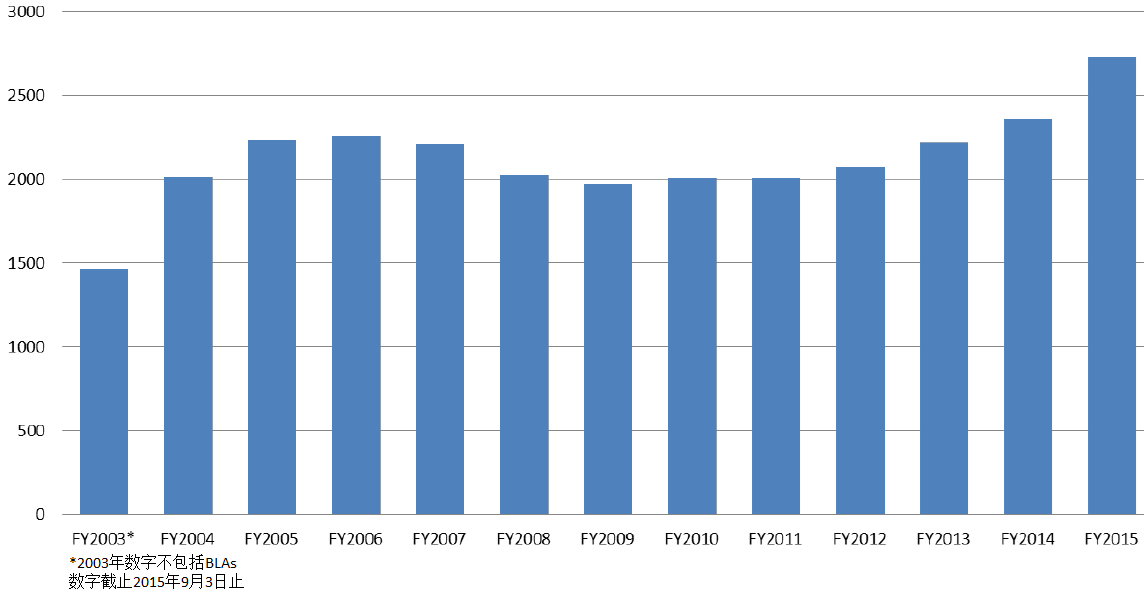
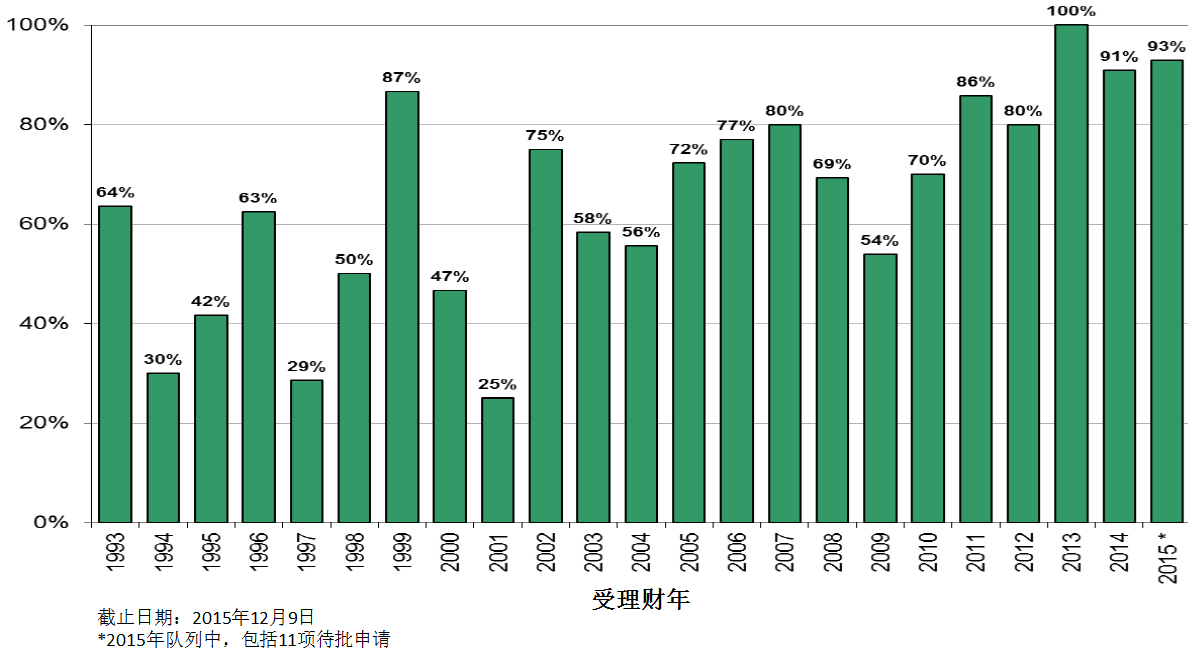
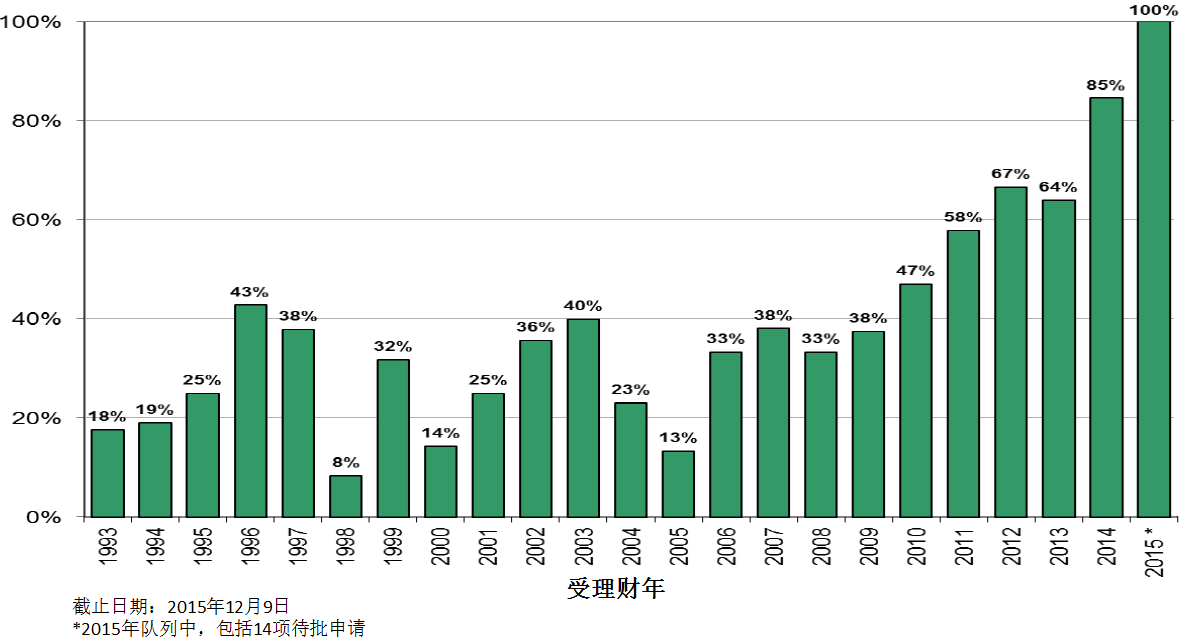
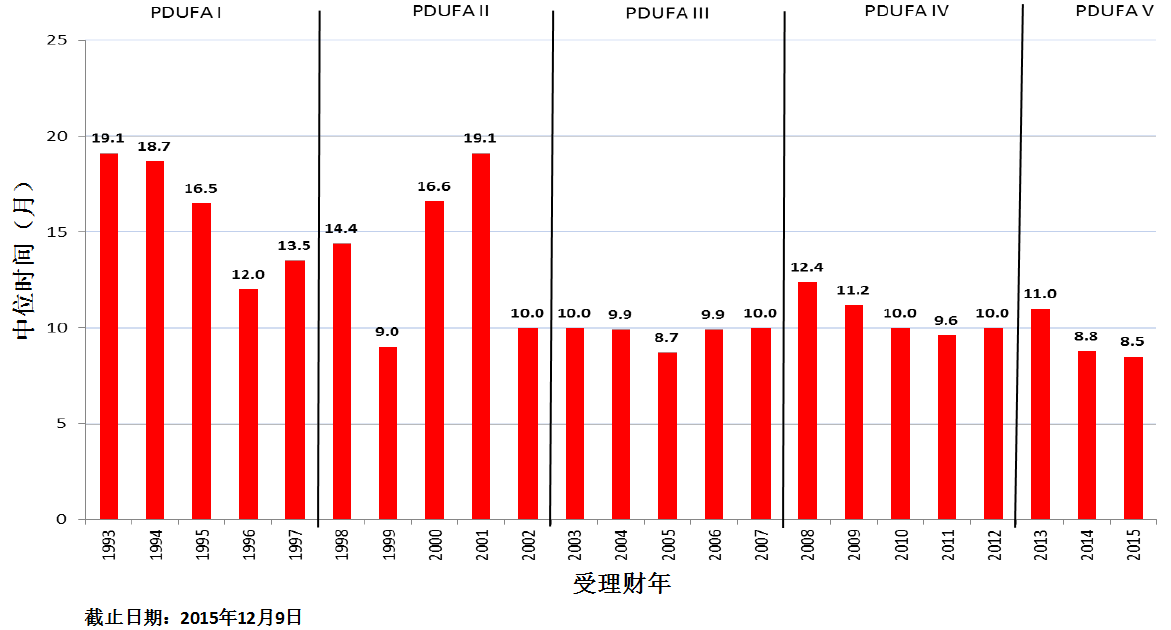
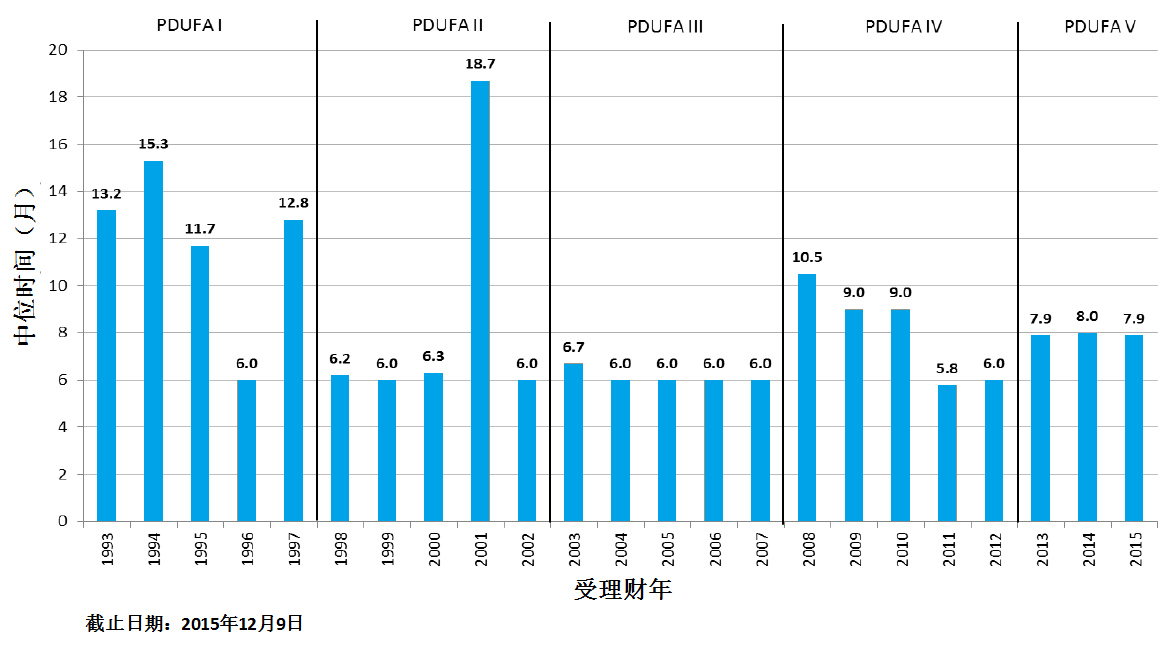
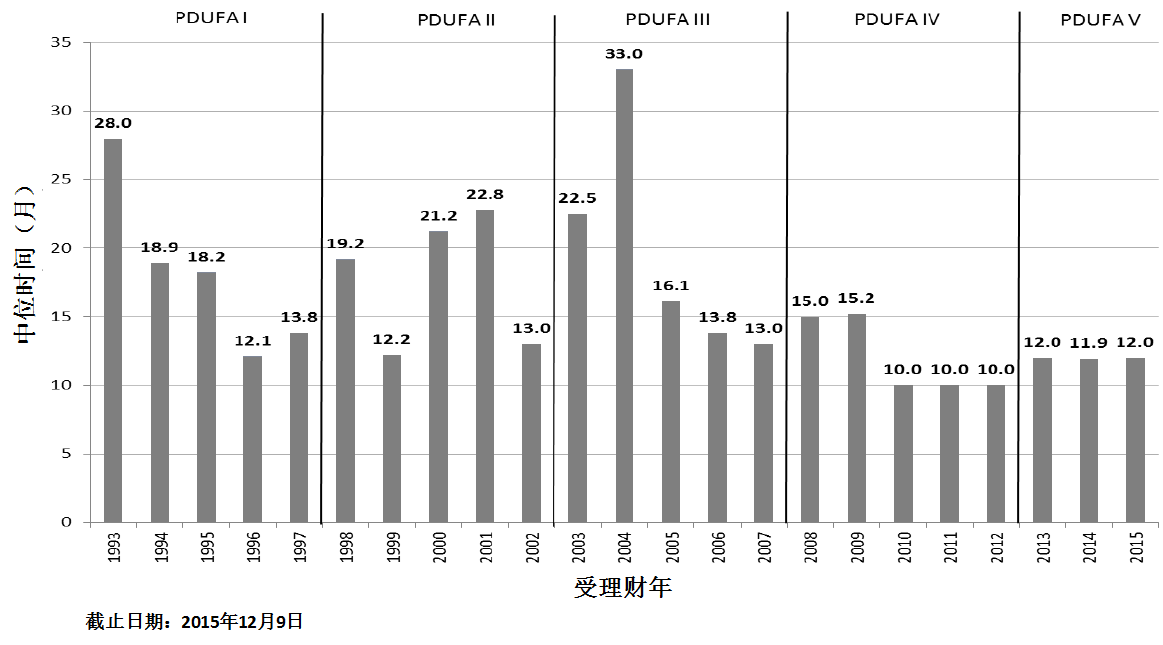
商业化IND研发管道保持强劲,绝大部分增长动力来自于生物制品。截止2015年12月9日,2015年CDER受理36份新分子实体申请;批准41个新分子实体,其中包括19个罕见病用药。首轮获批率创历史新高。由于新分子实体计划立卷审评需要额外增加时间,中位审评时间比预期略高。自2015年11月30日突破性治疗药物计划启动以来,CDER已经受理307份突破性治疗药物认定请求,给予95个突破性治疗药物认定,批准20个突破性治疗药物原始/补充申请。
表 1. CDER PDUFA V 审评绩效
2014财年
2015财年
申请提交类型
提交数
绩效(现行)
提交数
绩效(潜在)
优先审评通道 新分子实体NDA/原创BLA
24
96%
25
100%
标准审评通道 新分子实体NDA/原创BLA
14
93%
19
100%
优先审评通道 非新分子实体NDA/BLA
10
80%
8
100%
标准审评通道 非新分子实体NDA/BLA
72
97%
69
100%
第I类 NDA/BLA重新提交
6
100%
6
100%
第II类 NDA/BLA重新提交
32
97%
35
100%
优先审评通道 有效性补充申请
40
100%
37
98%
标准审评通道 有效性补充申请
146
90%
94
100%
第I类有效性补充申请
7
100%
0
--
第II类有效性补充申请
8
88%
7
100%
之前批准申请的生产补充申请
542
93%
455
94%
已实施待批(CBE)的生产补充申请
1017
95%
1017
97%
截止日期:2015年9月30日
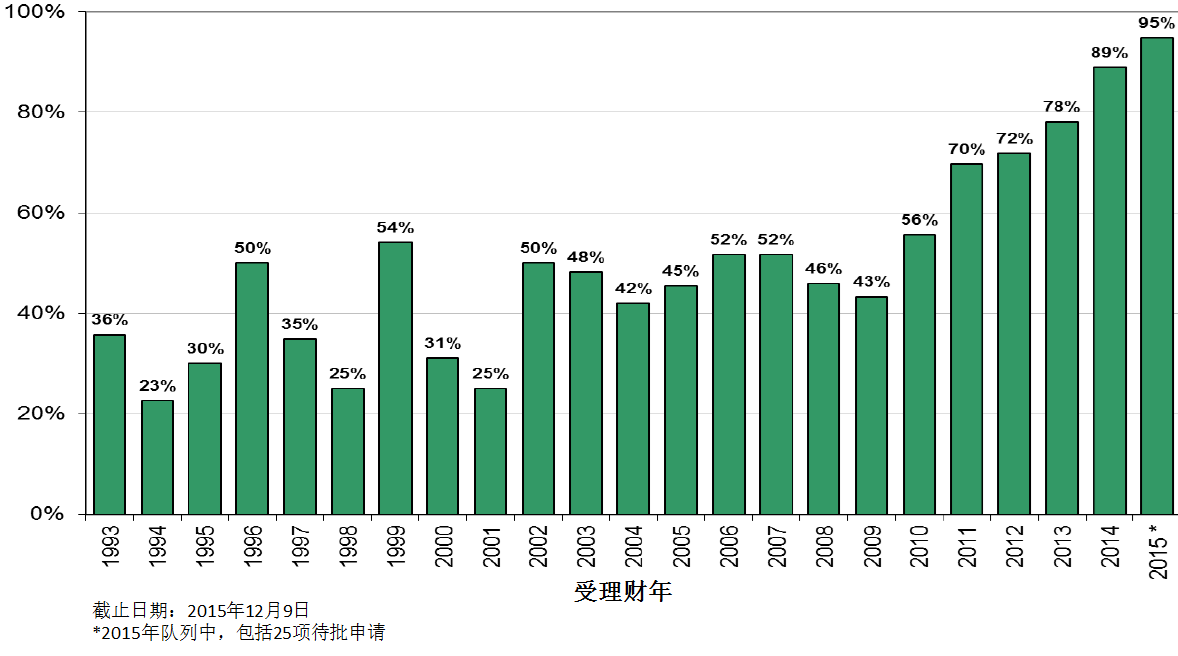
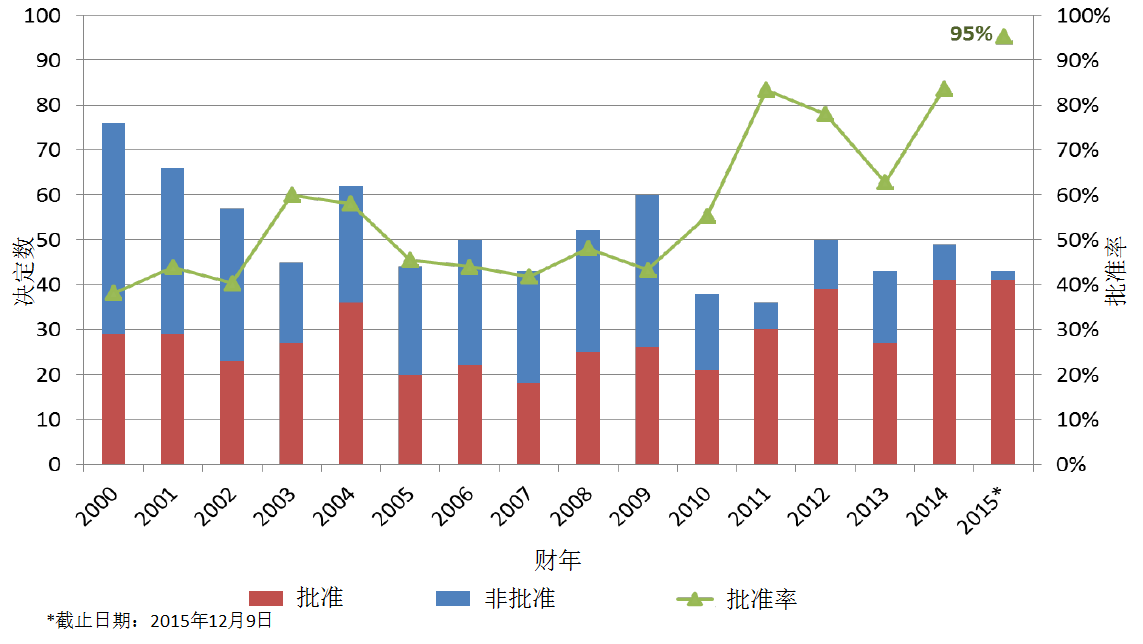
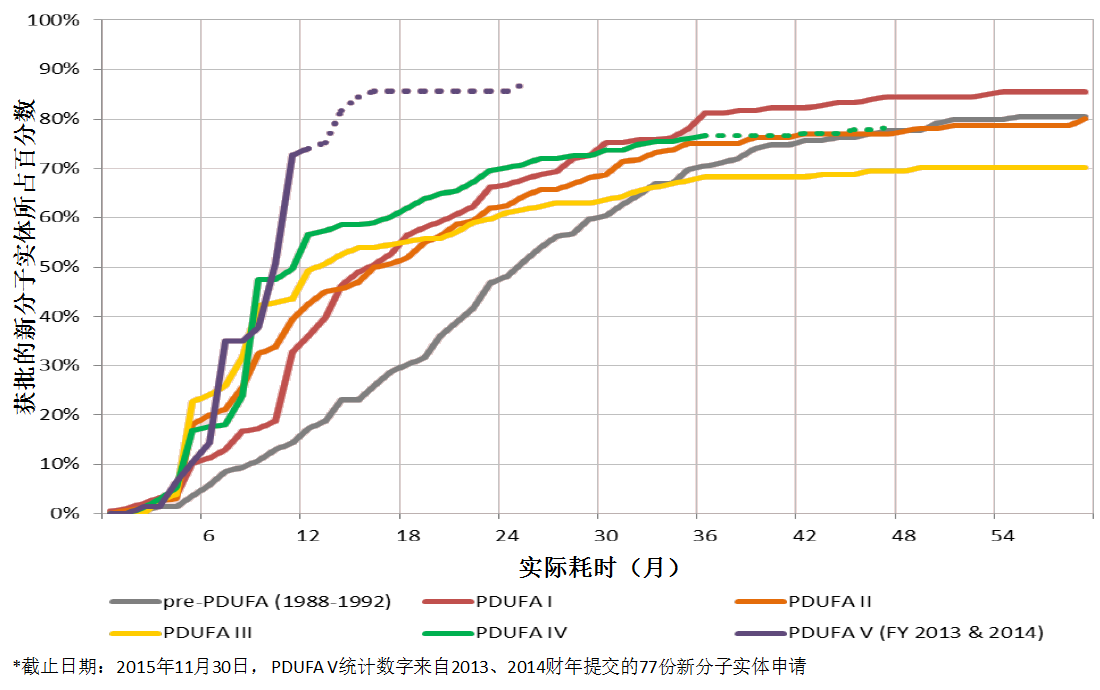
新分子实体首轮批准率被认为是一项重要的衡量指标。首轮批准率创下历史新高,并非由于CDER对法定标准的解读有所改变。
首轮批准率不断改善的原因包括,
FDA行业指南、IND期间召开会议澄清对于研发计划的期望,有助于提高新药申请/生物制品许可申请的质量。
新分子实体计划的实施,在立卷时提交完整申请,并与申请人更多的互动时间以解决缺陷。
靶向治疗药品针对经过选择的患者,获益更高,风险更少。
批准的药品中有更多的罕见病用药,使得获益/风险平衡有所改变。
对于申请人和FDA而言,突破性治疗药物认定起到了参与者各就各位的作用。
申请人的关注点从“跟随式创新”药品和存在较少有利的获益/风险平衡的多种治疗药品选择的疾病转移。
但从公共卫生的角度来看,首轮批准率创下新高并不一定意味着好的产出。
表 2. 2015年获批新药/生物制剂PDUFA绩效和审评通道
商品名
符合PDUFA绩效目标
首轮获批
优先审评通道
快速通道
首创药
在美国率先获批
加速审评通道
罕见病用药
突破性治疗药物认定
抗传染性疾病资质
SAVAYSA
COSENTYX
NATPARA
IBRANCE
LENVIMA
FARYDAK
AVYCAZ
CRESEMBA
UNITUXIN
CHOLBAM
CORLANOR
KYBELLA
VIBERZI
KENGREAL
ORKAMBI
ENTRESTO
REXULTI
PRALUENT
ODOMZO
DAKLINZA
ADDYI
REPATHA
VARUBI
XURIDEN
VRAYLAR
LONSURF
TRESIBA
ARISTADA
PRAXBIND
VELTASSA
YONDELIS
STRENSIQ
NUCALA
GENVOYA
COTELLIC
TAGRISSO
DARZALEX
NINLARO
PORTRAZZA
EMPLICITI
KANUMA
截止日期:2015年12月9日REPATHA在提出申请时有两个适应症,其中只有一个适应症涉及罕见病用药。
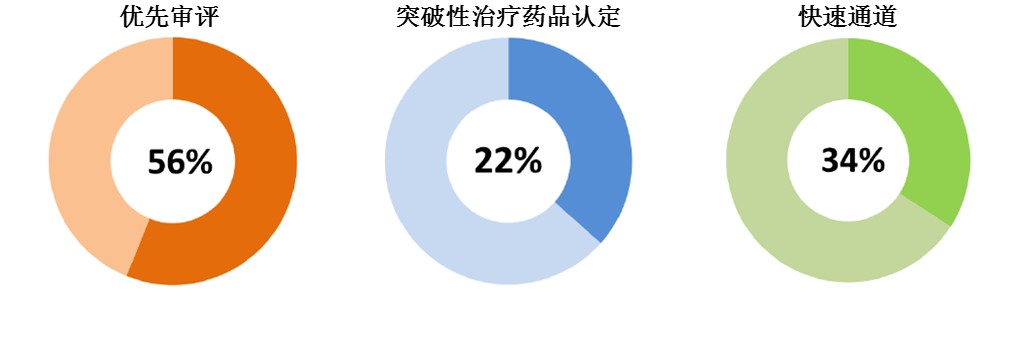
CDER确保新药接受加速审评。2015年获批的新药中,超过一半(56%)为优先审评通道审评;近1/4(22%)为突破性治疗药品认定;约1/3为快速通道审评。
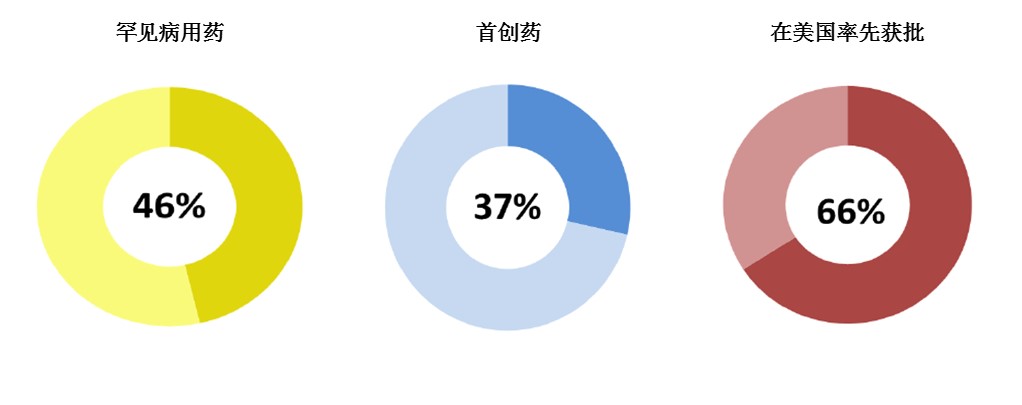
2015年,药物创新维持强劲势头。2015年获批的新药中,近一半(46%)为罕见病用药;超过1/3(37%)为首创药; 2/3(66%)为在美国率先获批。
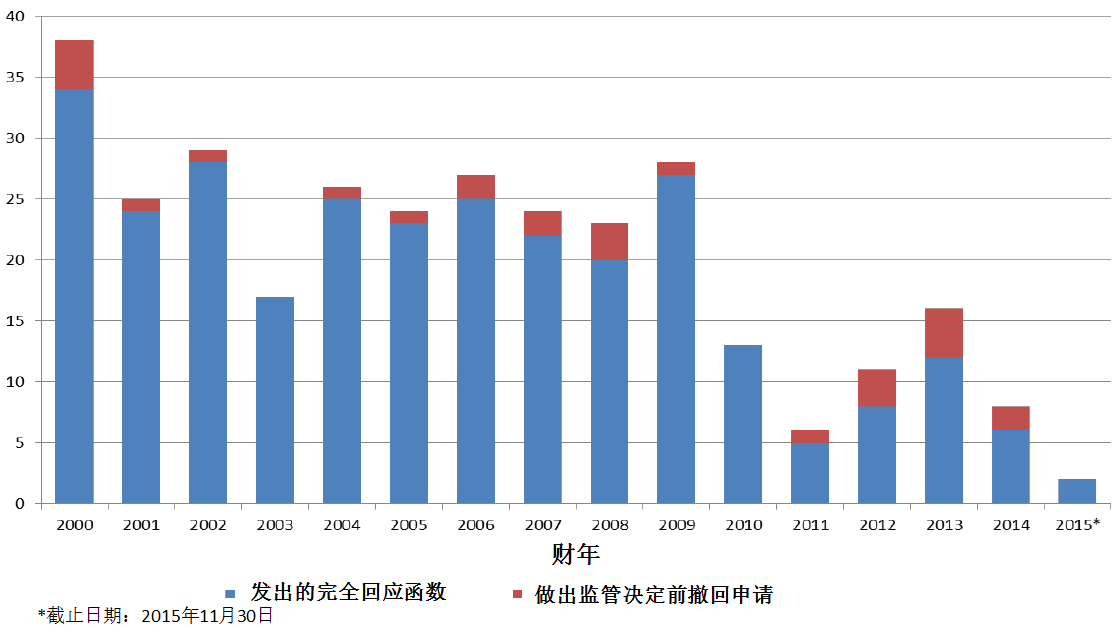
二.对新药研发和审评产生影响的PDUFA V/FDASIA计划
PDUFA V/FDASIA中涉及新分子实体和原创生物制品许可申请的审评内容:
力促申请提交前会议。
提交时申请完整;如果不完整,必须列入拒收。
74天信函―计划中的审评时间线,内部中期会议计划日期,需要召开咨询委员会的初步计划,就缺陷/资料要求早期展开沟通。
中期交流
在内部中期会议两周之内。
就确定的日期/资料请求、风险管理/风险评价与降低测量(REMS)、提议的晚期会议时间、咨询委员会计划更新等重要议题沟通、磋商。
专业审评信函 — 按照专业总结初步认定结果/缺陷
晚期会议 ― 关注信息共享、咨询委员会计划,并为余下的审评工作作出计划。
表3. 2013-1015财年对新药/生物制品审评产生影响的PDUFA/FDASA计划累计实施情况
2013财年
2014财年
2015财年
2015财年(截至2015年11月30日)
申请前会议
42
96
137
141
受理
53 106 166 174
拒收
2
3
4
4
第74天会议
44
98
157
163
中期沟通交流
33
80
135
150
专业审评函
5
8
10
10
晚期会议
17
64
111
121
首轮决定
6 64 115 133
决定后面谈
6 106 196 211
受理的重大修订
3 18 35 37
修订
166
2684
4740
5263
(根据2015年12月14日FDA CDER新药办公室主任J. K. Jenkins博士在2015 CMS/FDA Summit上的报告整理、翻译,演讲幻灯片
整理:识林-Kapok® www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com