首页
>
资讯
>
EMA 首次认证动物试验虚拟对照组,分享审评报告和 SOP
出自识林
EMA 首次认证动物试验虚拟对照组,分享审评报告和 SOP
2026-04-14
EMA于3月31日发布了一份认证意见草案 ,认定在大鼠剂量范围探索研究中使用虚拟对照组(virtual control group,VCG)的可行性,旨在减少或完全替代传统非临床研究中的动物使用。
正如2025年3月EMA认证了全球首个AI工具用于临床试验结果评价 ,这次EMA首次认证新方法学(NAM),对于动物试验替代具有里程碑意义。44页的认证意见包含大量评估细节,将成为药企和NAM供应商的权威实践参考,为未来类似应用建立了范本。
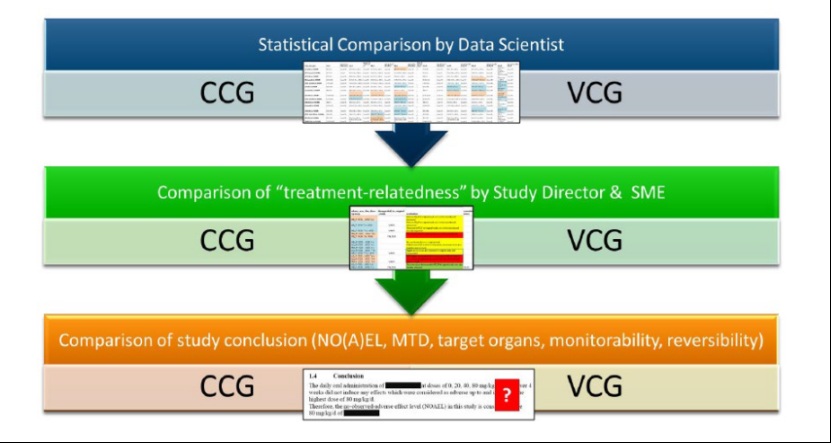
传统毒理学 研究中,约25%的动物被分配至对照组,主要是为实验提供生理基准,但这往往是重复产出标准化品系已知数据。依托“开发和实施虚拟对照组以减少毒理学研究中的动物使用”项目(VICT3R)数据库,VCG基于历史对照数据(historical control data,HCD)进行构建。VCG的建立过程包括对照数据的特征化描述,并为受试动物精准匹配在物种、品系、给药途径 及初始体重上高度一致的“虚拟孪生”。这一过程遵循标准操作规程(SOP) ,采用统计学方法并辅以专家判断。EMA强调,拟采用VCG方法的上市许可 申请人必须充分论证该方法不会影响研究结果或人类安全性。
EMA与VICT3R联盟在同日发布了一份SOP《在临床前研究中实施虚拟对照组的标准操作规程》 。该SOP详细阐述了临床前研究中VCG的生成与应用流程,涵盖HCD的收集、数据标准化、匹配标准选择(包括物种、品系、给药途径、初始体重等变量)以及VCG构建方法。
在剂量范围探索研究中,应用VCG的目标是通过确立未见不良反应剂量(NOAEL) 并初步识别不良发现,为后续关键性重复剂量研究的剂量选择提供依据。EMA从较早期阶段的具体场景入手促进NAM,较为务实。
EMA还将继续通过包括认证在内的多种监管机制和路径逐步推进NAM,确保人类与动物安全的前提下减少对动物试验的依赖。
动物试验替代和NAM推广近年来一直是全球药监的着力点,在2025年到2026年迎来多个里程碑。3月18日,FDA发布指南草案确立NAM验证框架 ,英国MHRA随后在3月25日发文提供非动物数据的提前审评机制(见今日副文 ),并明确此类数据的评估标准。
作者:识林-实木
责任编辑:识林-木姜子
识林® 版权所有,未经许可不得转载。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
非临床安全(Tox) :必读。需按SOP匹配VCG,验证历史数据兼容性,确保NOAEL判定与原始研究一致。 注册(Reg) :必读。关注EMA认证意见动态,在提交非临床资料时注明VCG使用依据。 研发(R&D) :必读。优化DRF研究设计,优先选择符合SOP匹配条件的实验参数。 数据管理(DM) :确保HCD符合CDISC SEND格式,建立内部数据标准化流程。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: 研发(非临床): 需将VCG匹配标准整合至研究方案设计,确保HCD选择与治疗组可比性。 QA: 监督VCG生成流程的GLP合规性,审核数据追溯文档。 统计分析师: 开发VCG专用统计模型,验证数据分布一致性(如预值分析)。 数据管理: 维护标准化HCD数据库(SEND格式),实施数据治理流程。 以上仅为部分要点,请阅读原文,深入理解监管要求。