报告的主要作者为 FDA 药品质量办公室(OPQ)的 Zhijin Chen。报告指出,原始 ANDA 的首轮批准率通常比较低,2018 财年至 2019 财年期间,仅 18% 的原始 ANDA 在首轮审评中获得批准。FDA 开展这一研究的目的是,收集和分析缓释片剂 ANDA 审评中提出的缺陷,以识别造成现在的低首轮批准率的根本原因。研究结果揭示了,大多数收到完全回应函(CR)的申请是由于多个审评学科不充分造成的。研究随后分析了在生产领域的常见缺陷。避免常见生产缺陷可以改善申请质量,从而改善仿制药申请的首轮批准率。
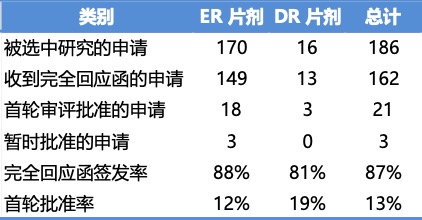
报告涵盖了 2017 年至 2019 年间提交的 186 件缓释片剂 ANDA 原始申请(其中 170 件 ER 片剂 ANDA,16 件 DR 片剂 ANDA)。研究发现,在这些申请中,首轮批准率为 13%,完全回应函发布率高达 87%。所研究的申请概览如下:
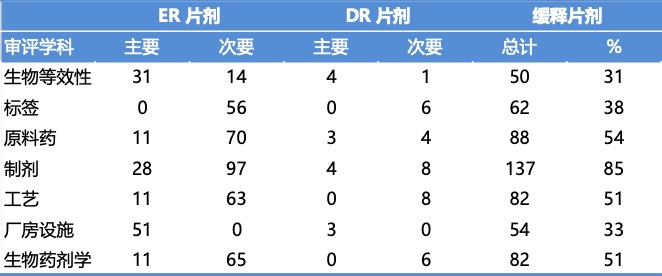
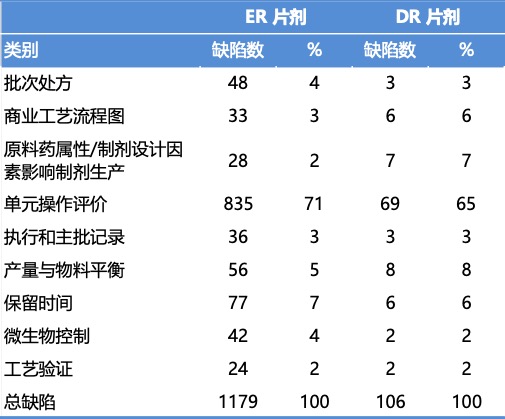
该研究随后归类并分析了生产相关缺陷。所研究的这些 ANDA 在首轮审评中共收到了 1,285 条生产相关缺陷,其中 275 条出现在完全回应函中。70% 的生产缺陷与单元操作相关。首轮审评中发现的生产工艺相关缺陷情况:
报告最后的结论指出,避免大多数常见生产缺陷可以改善申请质量,从而改善首轮批准率。
但前 FDA 仿制药办公室代理副主任,现 Lachman 咨询公司资深咨询师 Bob Pollock 先生评论指出,“实际上很多公司都在对缺陷进行分类和表征,并主动将这些知识纳入到新的申请提交中,以试图规避或减少缺陷从而避免多轮审评。但是多年来我接触过的几乎每家公司都反馈表明,即使采用这种积极主动的方法,不同的审评员也会在基本相同类型的产品上给出新的缺陷。这种‘移动靶’让申请人感到沮丧并且公司监管事务、生产和开发人员很难向高层做出解释。”
Pollock 继续表示,“我了解 FDA 正在寻找减少缺陷数量和提高仿制药首轮批准率的方法,但是当你看到新药申请(NDA)的首轮批准率超过 90% 时,新药和仿制药之间的脱节很难理解,尤其是不论新药还是仿制药,CMC 的大部分工作都是由药品质量办公室(OPQ)来完成的。我承认现在每年提交的 ANDA 数量大约在 700 到 900 件之间,而每年提交的 NDA 大约只有 90 到 120 件,这意味着有更多工作人员与新药申请人互动。而且 NDA 的审评涉及更多学科(包括临床毒理学、药理学等等),但是新药和仿制药申请人与 FDA 审评员之间的合作肯定有一些非常不同的差异,才导致体现在审评周期方面的巨大脱节。在 FDA 弄清楚之前,不要指望 ANDA 首轮批准率会有太大改善。也许 GDUFA III 的一些措施可能会有所帮助,但我个人总是会归结到审评一致性的问题上,而这是一个历史悠久的问题。”