|
首页
>
资讯
>
FDA 发2200封函提醒登记临床试验,如依法罚款可达千亿美元
出自识林
FDA 发2200封函提醒登记临床试验,如依法罚款可达千亿美元
2026-04-16
4月13日,FDA发布声明,称其已在3月30日向超过2200家医疗产品申办者及研究者发出提示函提醒其遵守向ClinicalTrials.gov提交特定临床试验结果信息的法定义务。该举措涉及超过3000项已注册的临床试验(其中部分为公共资助研究)。
FDA称其一项内部分析显示,在极可能属于强制报告义务范围内的研究中,有29.6%未向ClinicalTrials.gov提交任何结果信息。FDA局长Marty Makary批评某些申办者隐瞒不利的临床试验结果已成普遍现象。他也很关心临床医生能否获得该药物临床研究的全面数据,做出更合理的处方决策。
FDA指出,申办者及研究者常常不披露负面试验结果,导致公共记录存在显著缺口,并形成一种偏向于发表正面结果的“发布偏倚”(publication bias),使药物开发整体情况失真,表现为过度突出成功案例,而失败案例未被充分展示。此种信息缺口亦可能造成对医疗产品安全性与有效性的认知扭曲。
如此之多的不合规情形,其背景却是法律已明文要求临床数据公开。2007年通过的《FDA修正法案》(FDA Amendments Act)明确要求申办者在首名受试者入组后21天内,在政府数据库ClinicalTrials.gov上注册适用研究。试验的主要完成日期(primary completion date)后12个月内,申办者必须提交总结性结果信息(summary results)。
这次的提示函被FDA称为额外步骤(extra step)。其实FDA有权发送违规预先通知(Pre-Notices of Noncompliance)及违规通知(Notices of Noncompliance),但五年间仅发出8份。
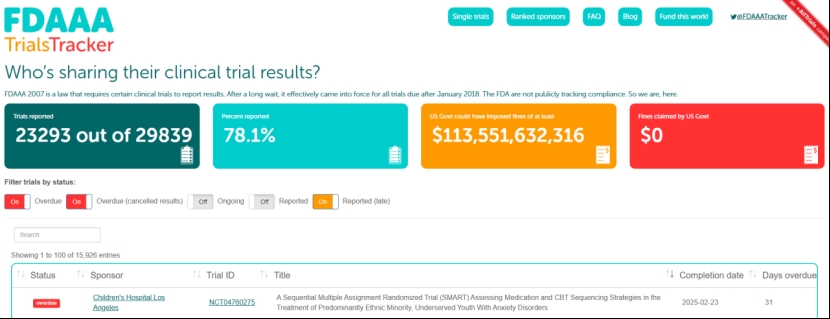
FDA也有权对违反临床试验报告义务的行为处以每日不超过10000美元的罚款(已发定稿指南)。然而,由英国研究人员创建的网站FDAAA Trials Tracker的数据显示,在超过29800项试验中,近22%未报告结果。该网站估计,FDA如果严格罚款,可能产生超过1135亿美元的巨额“收入”,但事实上FDA尚未开出任何罚单(见下图)。
无论是出于监管优先级还是成本考量,FDA对临床试验登记的管控确实较为“放松”。STAT援引业界人士言论表示,FDA此次行动更具象征性而非实质性,推测许多收到提示函的利益相关方可能会观望,等待实际的违规通知后再采取行动。识林曾有报道,罚款通知(最终还是未罚款)有效得多。
另外,尽管ClinicalTrials.gov仍是全球信息最为完整的临床试验登记平台,EMA在不断优化其登记平台CTIS,欧洲多国也在考虑替代方案。
作者:识林-实木
责任编辑:识林-木姜子
识林®版权所有,未经许可不得转载。
必读岗位建议: - RA(注册):了解FDA对处方药和医疗器械用户费项目的修订和延期,以及对药品上市后安全监管的增强措施。
- QA(质量管理):关注FDA对药品上市后安全性的监管增强措施,确保企业合规。
- 研发:了解儿科药品研究和医疗设备安全改进的相关要求,指导研发工作。
适用范围:
本文适用于美国境内的化学药、生物制品、疫苗、中药等药品类型,涉及创新药、仿制药、生物类似药、原料药等注册分类。适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。 文件要点总结: - 用户费项目修订:明确了对处方药和医疗器械用户费项目的修订和延期,以支持FDA的监管工作。
- 儿科药品研究:特别强调了儿科药品研究的重要性,通过《儿科医疗设备安全和改进法案》和《儿科研究公平法案》来增强儿科药品的安全性和有效性。
- 上市后药品安全监管:通过《增强药品上市后安全监管法案》增强了FDA对药品上市后安全性的监管权力。
- 临床试验数据库:规定了临床试验数据库的建立和维护,以提高临床试验的透明度和可访问性。
- 食品和药品安全:涵盖了食品和药品安全的相关条款,以保护公众健康。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |