|
首页
>
资讯
>
PMDA 阐明如何利用“既定条件”预先设定变更级别
出自识林
PMDA 阐明如何利用“既定条件”预先设定变更级别
2026-04-08
3月24日,PMDA发布《关于预先设定被认定为既定条件的药品(化学品)生产工艺参数的变更分类的基本考虑要点(早期考虑)》,旨在澄清化学药生产过程中被判定为既定条件(Established Condition, EC)的工艺参数如何在初始申报时预设变更分级。
在EC的具体操作方面,FDA的《ICH Q12: FDA 监管产品的实施注意事项》将EC相关变更分级的主动权(也包括责任)交给企业,未作更具体指导;EMA的《在欧盟实施ICH Q12药品生命周期管理的技术和监管考虑的说明》甚至指出Q12中的EC评估框架与其变更法律框架不符,并要求EC的使用遵循其变更法规。我国未对EC发文,目前主要聚焦于批准后变更管理方案(PACMP)工具。曾有文献指出,日本对于EC的应用较为广泛。
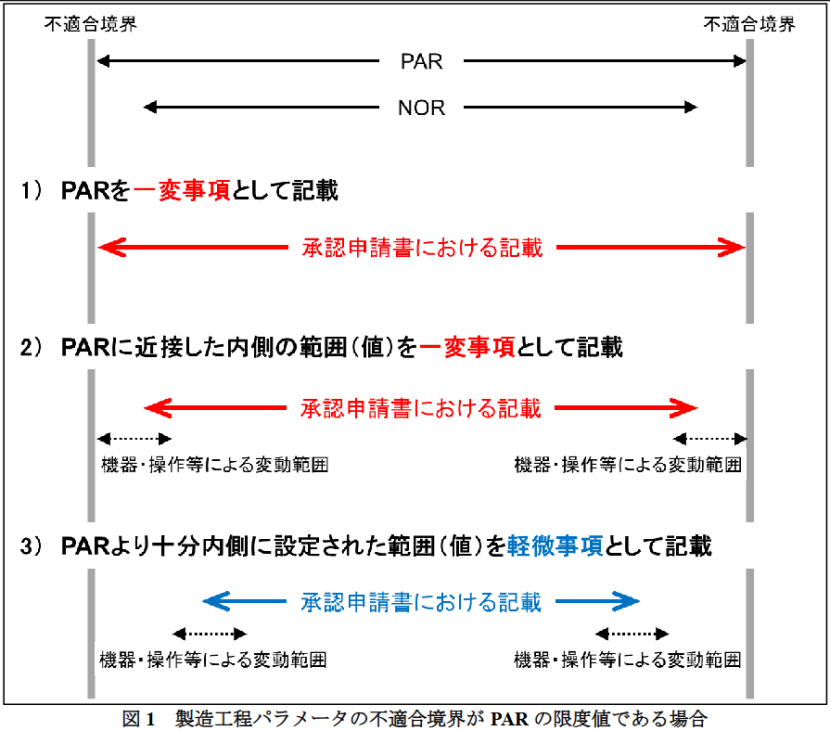
PMDA指南侧重于厘清可接受范围(Proven Acceptable Range, PAR)与操作范围(Normal Operating Range, NOR)的关系,旨在明确“一部变更”与“轻微变更”事项的界限(大致可类比我国“重大变更”和“中等/微小变更”)。此外,指南点明工艺能力指数(Cp/Cpk)作为判定“变更范围余量”的客观统计依据,为企业提供了一套比欧美指南更具参考性的模板。
指南将参数与“不适合边界”的关系分为三种情况讨论。其中,“不适合边界”即工艺参数的上限范围,有数据证实超出则产品不合格,下面将采用ICH Q8的术语“失败边缘”(edge of failure)。
情形一:当PAR限度=“失败边缘”时
当申报工艺参数波动超出PAR即导致质量不合格时,“失败边缘”等同于PAR限度。申报资料提交方式分为三种策略:
- 申报工艺参数范围=PAR限度:此时变更工艺参数均为“一部变更”。
- 申报工艺参数范围<PAR限度,与NOR相当:此时即使参数变更范围还在PAR内,变更仍需按“一部变更”处理。申请人需考虑设备性能及操作变动对参数波动的影响。
- 申报工艺参数范围远小于PAR限度:若申报范围与PAR限度值之间保有充分余量,后续工艺参数变更可按“轻微变更”处理。
PMDA强调,申请人在申请“轻微变更”分级时,必须提供科学客观依据,避免定性描述。客观依据包括工艺验证结果、基于实际设备生产数据的工艺能力指数(Cp/Cpk)以及设备校准与验证记录。
情形二:当“失败边缘”未确定,但工艺参数被定为CPP时
对于关键工艺参数(CPP),即使在研究范围内未触及“失败边缘”,PMDA也推定“失败边缘”存在于PAR限度值附近的外部。因此,工艺参数的变更分级设定原则沿用情形一。
情形三:当“失败边缘”未确定,工艺参数也非CPP时
若并未触及“失败边缘”,申报工艺参数非CPP,但无法合理排除对质量的影响而被判定为EC,申请人可将PAR范围内的工艺参数变更均作为“轻微变更”处理。此时申请人还需在CTD中解释该参数对关键质量属性(CQA)影响有限的理由及研究结果。
不过,上述操作都是基于单个工艺参数范围。PMDA补充强调,工艺参数间存在显著相互作用的情况,此时单纯设定个体参数的PAR不足以保证质量。申请人需通过实验设计(DoE)等多变量分析,以多变量控制、模型或设计空间(Design Space)的形式申报。此类EC的变更分级判定准用本指南基本思路,并依据模型变更带来的风险程度进行个案处理。
最后,PMDA提醒企业,申报资料中变更分级在产品上市后并非就此不变。在产品生命周期管理中,若因生产场地变更或设备调整导致原有的“安全余量”缩减,企业必须通过变更管理程序,主动将“轻微变更”事项调回“一部变更”。
作者:识林-实木
责任编辑:识林-木姜子
识林®版权所有,未经许可不得转载。
适用岗位: - QA(质量保证):必读。建议深入理解ICH Q12指南中关于已建立条件(ECs)的识别和管理,以及如何与FDA的监管框架相结合。
- 注册:必读。建议熟悉ICH Q12指南中关于药品生命周期管理的监管工具和原则,以便在注册申请中正确应用。
- 研发:必读。建议了解ICH Q12指南对于药品研发过程中CMC变更管理的影响,特别是在提出ECs时的科学依据。
- 生产:必读。建议掌握ICH Q12指南中关于生产过程中变更管理的要求,以及如何在生产质量体系(PQS)中实施。
适用范围:
本文适用于FDA监管的化学药品、生物制品、疫苗、中药等药品类型,包括创新药、仿制药、生物类似药、原料药等注册分类。适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 要点总结: - 已建立条件(ECs)的管理:强调了在提交原始NDA、BLA或ANDA时,申请人应明确提出特定的ECs及其变更的报告类别。
- 变更管理协议:提出了通过补充申请或后批准变更管理协议(PACMP)来添加、消除或修改已批准的ECs。
- 药品生命周期管理文件(PLCM):建议在eCTD部分3.2.R中以表格形式提供PLCM文件,包括提出的ECs、变更报告类别、可比性协议列表和后批准CMC承诺。
- 药品质量体系(PQS):强调了PQS在支持ICH Q12工具使用中的重要性,并指出FDA将通过常规检查和其他信息来评估PQS的有效性。
- 监管评估与检查的关系:明确了ICH Q12工具的使用不会改变FDA对申请信息的评估或设施检查的流程。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读:ICH Q8 Pharmaceutical Development适用岗位(必读)- 研发(R&D):深入理解药品开发过程中的科学方法和质量风险管理。
- 质量管理(QA):确保药品开发符合ICH Q8指南要求,建立设计空间和控制策略。
- 注册(Regulatory Affairs):在药品注册文件中准确呈现药品开发信息,包括设计空间和控制策略。
工作建议- 研发(R&D):应用科学方法和质量风险管理来设计药品及其制造过程,确保产品质量。
- 质量管理(QA):监督药品开发过程中的质量控制,确保设计空间和控制策略得到有效实施。
- 注册(Regulatory Affairs):在CTD格式的注册文件中,合理布局药品开发相关信息,确保监管机构能够清晰理解。
适用范围本文适用于化学药品、生物制品、原料药等多种药品类型,包括创新药、仿制药、生物类似药等注册分类。适用于跨国药企、大型药企、Biotech等不同企业类别。发布机构包括中国、美国、欧盟等ICH成员国。 要点总结- 设计空间(Design Space):提出了设计空间的概念,强调了在设计空间内操作不视为变更,超出设计空间则需要启动监管变更程序。
- 质量风险管理(Quality Risk Management):强调了质量风险管理在药品开发过程中的重要性,特别是在识别和控制对产品质量有影响的关键因素。
- 关键质量属性(Critical Quality Attributes, CQAs):明确了CQAs的识别和控制是确保产品质量的关键步骤。
- 控制策略(Control Strategy):提出了基于对产品和过程深入理解的控制策略,包括对关键过程参数和物料属性的控制。
- 生命周期管理(Lifecycle Management):强调了在整个产品生命周期中持续改进和创新的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 【文件概要】
该文件针对药品(化学品)生产过程中被认定为既定条件(Established Condition, EC)的工艺参数变更分类提出预先设定的基本考量框架。文件基于ICH Q12指南原则,重点分析已确立条件(EC)与变更分类(部分变更申请事项或轻微变更报备事项)的关联性,强调通过风险评价(包括工艺理解度、设备特性、控制难度等)确定变更管理策略。核心内容涵盖三类场景:(1)不适用边界与PAR(Proven Acceptable Range)限度值重合时,需将PAR或其内测范围作为变更分类依据;(2)未明确不适用边界但属于关键工艺参数(CPP)时,参照PAR外推风险采取保守策略;(3)非CPP但可能影响产品质量的参数,可归类为轻微变更。文件同时指出变更分类需在生命周期中动态调整,例如因生产设备更新或长期数据积累导致风险变化时需重新评估。 【适用范围】
本文适用于日本境内化学药品生产企业,涵盖创新药与仿制药的工艺参数变更管理,涉及ICH Q12框架下的EC认定及后续变更分类设定。适用对象包括大型药企、跨国药企及CDMO等需向PMDA提交变更申请的实体。 【影响评估】
本文要求企业强化对生产工艺参数的科学研究与风险管理,可能增加早期研发阶段的数据生成成本,但通过预先设定变更分类可加速后续变更审批流程。对依赖快速工艺优化的Biotech企业及CDMO的合规管理影响显著。 【实施建议】 - 必读岗位:
- CMC:需结合PAR与NOR数据,在申报资料中明确参数范围及变更分类的科学依据。
- QA:建立动态风险评估机制,监控生产数据以触发变更分类再评估。
- 注册:在CTD模块2.3/3中论证变更分类合理性,重点关注CPP与非CPP的区分逻辑。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |