首页
>
资讯
>
【识林社区】优质问答集锦3
出自识林
2022-04-30
以下是识林社区中的部分优质问答:
1.【研发】ICH M7一类杂质可摄入量计算:用动物实验的TD50 推导出人类致癌剂量的背后逻辑是什么?
ICH M7 中原文:根据致癌性数据库(CPDB数据库官方名称),环氧乙烷的TD50 为21.3 mg/kg体重/天(大鼠)和63.7 mg/kg体重/天(小鼠)。在计算可接受摄入量时,采用了剂量较低的大鼠值(即更保守)。
为推导十万分之一致癌率的剂量,将该值除以50000:
21.3 mg/kg÷50,000=0.42 µg/kg.
推导人类每日总摄入量:
0.42 µg/kg/日×50 kg体重=21.3 µg/人/天。
从以上计算看出,对不同物种(大鼠和人)在终生服用的情况下,如果每天每Kg体重摄入量相同时,各自致癌的风险率就相同,但是大鼠在TD50 中实验按照2年最寿命,M7规定人类按照70年。所以这样的话他们的每Kg体重的终生累积服用剂量就相差甚远(35倍!)。这点和指南中指出的Haber法则:认为浓度(C)×时长(T)=常数(k)似乎是相违背的。
我还设想过,是否因为不同物种致癌的敏感度不同,如果是这样的话就多了一个未知系数(实验动物与人类的敏感度比值),所以会导致无法用动物实验的TD50 推导出人类致癌剂量,所以也说不通。
毒理学 门外汉,请大佬指教。
社区用户@南瓜:
以下是我查到的信息和一些个人理解,非毒理专业人士,供参考:
ICH M7 中注释4从 TD50 线性外推的示例中有如下描述:“可以根据啮齿类动物致癌效价数据,例如 TD50 值(导致50%肿瘤发生率的给药剂量相当于患癌风险可能性水平为1:2),来计算特定化合物的可接受摄入量 。通过简单地将 TD50 值除以50000来线性外推至十万分之一的发生率(即可接受的终生风险水平)。该方法类似于 TTC 使用的推导方法。 ”
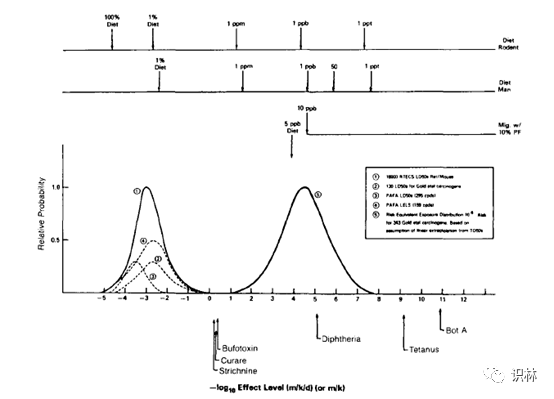
既然“该方法类似于 TTC 使用的推导方法”,那么我们就来看一下 TTC 是怎么推导出来的:1984年 Peto 和 Gold 等人发表文章[1] [2]建立了CPDB数据库,并首次定义了"TD50 "这一用来数值化描述致癌效力(Carcinogenic Potency)的指标,其定义如下"that dose rate (in mg/kg body weight/day) which, if administered chronically for the standard lifespan of the species, will halve the probability of remaining tumorless throughout that period."。Rulis(时任FDA食品安全与营养中心)等人分别于1986、1987、1989年发表文章[3] [4] [5]基于 CPDB 数据库中343种致癌物数据通过线性外推将致癌效力分布转换为了风险等效暴露-概率分布图(见图1),从而确定了在10-6终生风险水平下50ppb即0.15μg/人/天的建议阈值。1990年 Munro 等人[6]确认了这一研究,1993年 FDA 基于此研究确定了10-5终生风险水平下1.5μg/人/天的监管阈值(ToR)[7],也就是目前TTC的来源。ICH M7 注释4中的方法同样来源于此,只不过 TTC 是根据几百种致癌物的外推数据通过统计而来,注释4这个方法是根据某化合物的特定 TD50 外推而来。
在 Rulis 等人的文章中对于致癌效力分布的转换过程中有一个假设"using the simple assumption that low-dose risk equals potency times exposure (where, for the purposes of this discussion, potency is taken to be the slope of a straight line connecting zero risk/zero exposure to a point corresponding to a risk of 0.5 at the TD50 dose of Gold et al",也就是风险=效力* 暴露,其中效力取自(TD50 ,0.5)与0点所作直线的斜率。所以,我的理解这个假设就是简单的认为在动物和人之间每单位剂量率导致的终生致癌风险是一致。那么这个假设有什么依据呢?
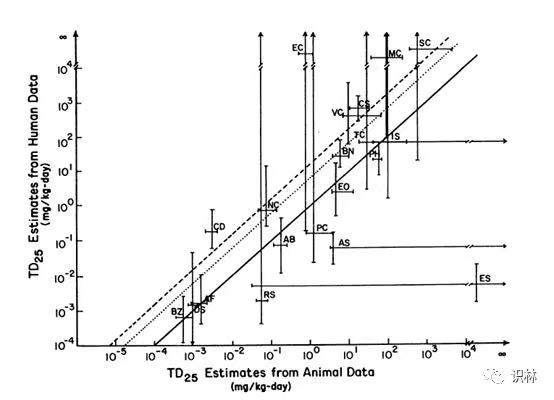
其实在1984年 Peto 和 Gold 等人定义 TD50 这一致癌效力指标之前或之后,有很多人都提出了致癌效力的数值化指标,只不过可能 TD50 和 LD50 更为相似,所以普遍被大家接受。所以关于物种间致癌效力转换的讨论在这个方法建立前后一直都存在,几十年来有大量的文章都讨论了这一话题,结论各不相同。其中在 TD50 建立前后也有部分研究(比如1979年 Crouch 等人[8],1988年 Allen 等人[9],1991年 Goodman 等人[10],1992年 Gold 等人[11])得出了人和啮齿动物之前的致癌效力是趋同的。比如1991年Goodman 等人的文章中定义了 KhR 作为从啮齿动物到人的致癌效力转换因子(其中大鼠到人为 Khr,小鼠到人为 Khm),该文章提到 FDA 的决策者认为Khr=Khm=1,而 EPA(美国环保署)的决策者认为 Khr=5.9,Khm=,13。Goodman 等人通过对 Allen 等人的研究数据[9]进行再次分析后认为(见图2 实线1,点线5.9,虚线13),EPA 将啮齿动物到人的致癌效力转换因子高估了,更认可 KhR≈1的结论。
但是动物和人之间的致癌效力究竟怎么换算这是一个很难说的清的问题,对于你问题中关于人和动物寿命不同的疑惑,可能 Goodman 文章中的一段对于致癌的生物学机理的讨论可以给你一些启示:“假设人类和小鼠的所有组织在例如受到电离辐射的均匀轰击时都同样可能发生肿瘤。因此,人类在任何给定时间段内患肿瘤的概率都比小鼠大(仅仅因为有更多的组织),比率为 Mh/Mm,Mh 和 Mm 分别代表人类和小鼠的质量。现在众所周知,癌症的发病率会随着年龄的增长而增加,时间的幂次方取决于恶性肿瘤的类型。让我们假设是4次幂, 那么在小鼠的一生 Tm 中发生肿瘤的概率比在人类一生 Th 期间发生肿瘤的概率小,即 (Tm/Th)4。 因此,我们预计小鼠辐射诱发癌症的终生发病率将比人类低的因子约为:(Mm/Mh)(Tm/Th)4 = (30 g/70 kg)(2 年/70 年)4 = 1/109。然而有证据表明,辐射诱发癌症的小鼠/人类实际比例更接近一致。 我们都知道这个悖论的答案:这是因为导致致癌的生化和生理事件在小鼠中比在人类中进行得更快。较小动物的细胞分裂速度更快。小鼠的心输出量是每分钟总血量的 100%,而人的心输出量仅为每分钟血量的 5%。这些以及其他因素,无论是已知的还是未知的,都会导致肿瘤诱导的不同概率和滞后时间,以及不同的剂量反应 。”[10] 所以,从这个角度来看,我们如果要弄清楚不同物种间的致癌效力的话也不能只考虑寿命的不同,对于某一特定化合物,所有关于其吸收速率和机制的差异、向靶器官的分布、生化转化为活性或非活性代谢物、清除和致癌机制等信息都应予以考虑。我认为您提到是否需要考虑寿命的换算可能对于同一物种同一化合物的致癌效力可能更为适合,就像 ICH M7 中提到的 LTL 暴露计算。
其实,ICH M7 推荐的简单线性外推这一方法是相当保守的,有以下几个方面:
(1)TD50 的计算过程中采用了很多保守的假设;
(2)线性外推采用了考虑最敏感的物种、部位、性别等因素的TD50 ;
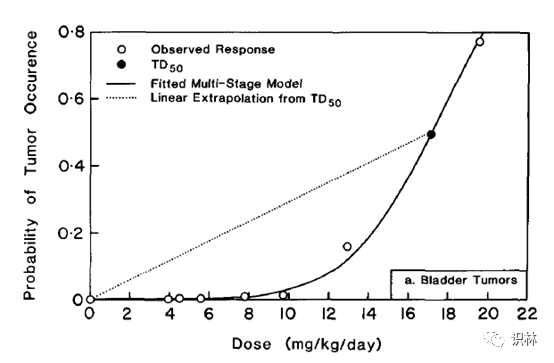
(3)在相同风险水平下,简单的直线线性外推要比一些其它的外推方法比如线性多阶段外推(LMS)的得出的剂量要低(见图3);[12]
(4)这种方法并没有考虑人类的防御机制而设置一个无剂量反应的阈值,通过外推得到的量极低对于某一化合物可能早已在这个阈值之下了;
希望这些对你理解这一方法“背后的逻辑”有帮助,其实这个方法是建立在很多假设的基础之上,可能并不需要那么精确的计算或换算。包括终生致癌风险10-5虽然是建立在统计数据的基础上,但最终的确定也是有一些主观成分的,认为这个风险足够可以接受。
如有错误或不足请指出,谢谢
社区用户@感方:
TD50 值线性外推是基于啮齿动物的最敏感部位的致癌性数据,通过除以50000线性外推至十万分之一的患癌概率;这个外推确实是没有考虑物种和器官的差异的,但不考虑也是更保守的结果,人类相对于啮齿动物而言,肯定剂量是更耐受的;
当然 M7 Note4 中也有描述:使用啮齿类动物致癌性研究中最保守的 TD50 值并不不考虑其与人类相关性的,作为该方法的替代方案,可以对已有致癌性数据进行深度毒理学专家评估,以便首先确定与人 类风险评估相关性最高的发现(例如,物种,器官),作为推导线性外推参考点的依据。
深度毒理学评估后,推导出线性外推的起点值,但明显会比较复杂。
因此,简单粗暴的 TD50 外推,除以50000,大家不爱吗? 毕竟外推算得的数据比保守的 TTC 通常要大的,在分析手段日新月异的今天,相信这不是难到我们制药人的关键。
2. 【注册】临床策略问题 补充申请还是NDA?
大佬指教。请问,一个境外生产药品,在境外获批适应症 为成人和儿童,在中国仅获批了儿童适应症。现在直接进行该品种成人适应症的上市申请(已取得沟通交流会认可),那么我这个产品现在应该是按照补充申请 变更 用药人群?还是按照增加境外已获批新适应症来报一个新的NDA 呢?
社区用户@Rlch:
大概率要按照NDA的途径来申请
补充申请途径
虽然《已上市化学药品和生物制品临床变更技术指导原则》 中提到:
“重大变更 A 类主要包括以下情形:
(1)已批准适应症的变更
①适用人群的变更,例如:在成人的基础上扩展至儿童等适用人群年龄范围扩大的情形。”
但是在适用范围中提到:“本指导原则适用于化学药品、预防用生物制品 和治疗用生物制品。对于已上市药品增加境内未批准的新适应症、改变给药途径 等,需按照药物临床试验 和上市许可 申请通道进行申报和审评审批。”
当然,这里没提适用人群的问题,最好还是有个实例。
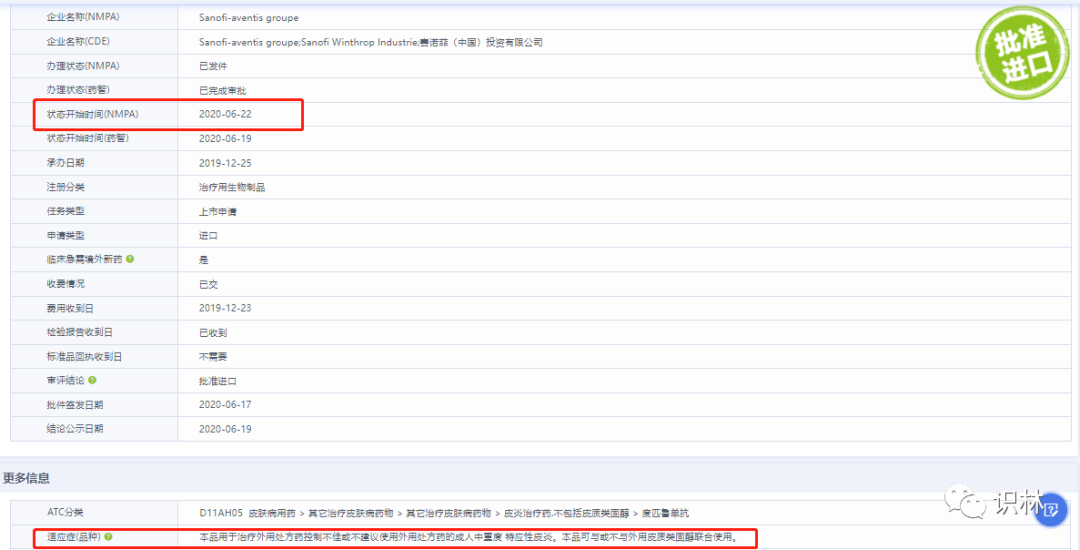
实例:度普利尤单抗 注射液(达必妥)-赛诺菲
2022年2月24日,赛诺菲达必妥的用药人群拓展得到了国家药品监督管理局的审批。
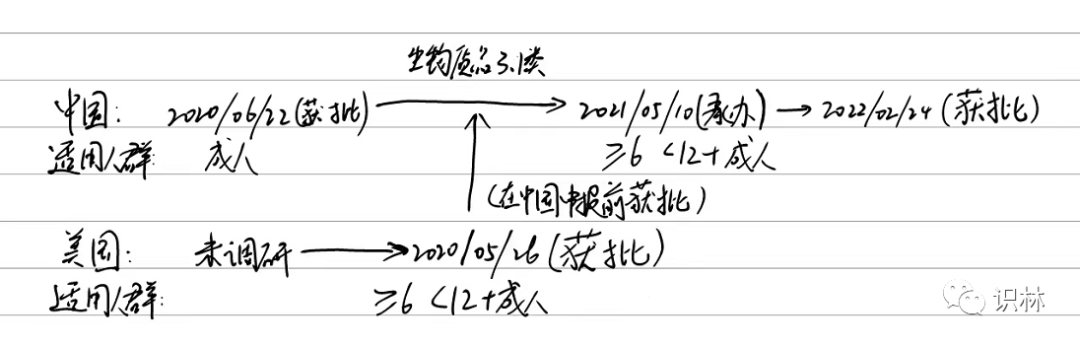
经调研,其获批路径是这样的:
2020年6月22日获批的适用人群是成人
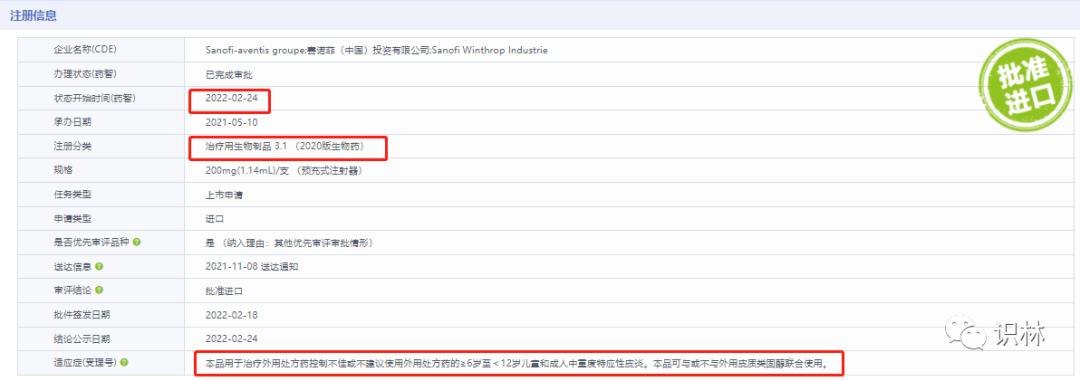
2022年2月24日获批的适用人群范围是大于等于6岁,小于12岁以及成人。其注册分类 是治疗用生物制品3.1类
根据《生物制品注册分类及申报资料要求》 ,生物制品注册分3.1类是:
“境外生产的境外已上市、境内未上市的生物制品申报上市”
这里的逻辑链条还缺最后一块,即在中国申报上市的时候,国外是否有已经批准的适用人群。经查询:6-11岁的适用人群,在2020年5月6日的时候,就在美国FDA已经获批。而在中国申报相应适用人群的时候,承办日期是2021年5月10日。
同时,中国只有赛诺菲的度普利尤单抗注射液在中国获批,也就是说,在申请拓展适用人群的时候,中国没有相同品种的相同适应症,相同适应人群获批。
综上所述,您的产品很有可能需要按照NDA 的途径申报。
画了张赛诺菲的申请时间图,希望可以帮助您理解:
3. 【生产】发酵周期延长或缩短的问题?
抗生素 发酵生产(180吨发酵罐)工艺规定:发酵周期>150h。实际生产中发酵周期基本为200h左右(效价11000u/ml左右),偶尔会延长到220h(效价12000u/ml左右)。但也会出现180h放罐情况(效价9000uml)。一般提前放罐批次基本都是染菌造成的,但是车间不走偏差 处理,每个月都会有3-5批次染菌。
1、提前放罐批(不显示染菌)效价低,批记录和正常放罐批次基本一致,无异常。但是年度回顾或持续工艺确认中, 效价会出现不良趋势,不好分析该情况。
2、常年发酵不染菌会不会被挑战?
社区用户@圣人有点冷:
回答:
从发酵周期来看是符合工艺要求的,发酵工艺控制不仅仅发酵周期,还包含DO、PH、搅拌速率以及培养基 添加等,判定的终点是结合目的产物例如产品的效价,效价有异常趋势,说明我们的工艺波动性较大,还是会回到发酵周期的问题上来,有经验的老师都会联想到染菌的问题。
偏差 的定义是偏离既定的程序或标准,如果你的体系文件里面规定了发酵过程不能染菌,那就需要按照偏差程序进行处理的。不管你的程序文件的规定如何,从工艺控制的角度还是建议进行调查分析。车间不走偏差处理是不可取的。首先应对染菌进行鉴定,分析可能的来源,并建立菌种库,便于后续的调查。调查可从人员资质和培训、设备 清洁消毒或灭菌、设备的密封性、物料的微生物限度 ,是否煮沸霉菌,液体或气体是否除菌过滤 等,人员接种的方法是适用,清洁消毒灭菌的方法是否经过验证,接种的环境是否符合要求,如果调查发现导致染菌的根本原因是相同或相似的,首先需分析导致重复发生的原因,是根本原因调查不彻底还是CAPA 不充分,如果重复发生三次以上建议升级该偏差,并将相应的原因和资源需求反映给高层管理者。如果放任不管,反应我们质量管理的体系是存在缺陷的,容易被官方审计时挑战。
4. 【质量】批生产记录上是否必须贴备料称量打印纸,打印纸日期是否是必须项?
普通固体制剂,备料称量后电子秤打印纸是否必须贴到批生产记录上,如果必须,是否可以不带时间
答案来自社区用户@李振华:
建议:
首先明确在制药过程中,所有与生产相关的记录,标签 等都是有明确格式、规定,也需要进行一定管理的。
关于普通固体制剂,备料称量后电子秤打印纸是必须贴到生产记录上(且是批生产记录的一部分),同时还需要相关人员签名确认,填上确认日期(操作人员,复核人员)。
然后打印的称量纸也需要打印日期,因电子秤打印纸是批生产记录 的一部分,相关数据应具有可追溯 性,或者国外经常说的要符合数据完整性要求。
依据:
中国版 GMP 第八章 文件管理 第一节原则 第一百六十条 应当尽可能采用生产和检验 设备自动打印的记录、图谱和曲线图等,并标明产品或样品的名称、批号和记录设备的信息,操作人应当签注姓名和日期
第四节 批生产记录 第一百七十四条 在生产过程中,进行每项操作时应当及时记录,操作结束后,应当由生产操作人员确认并签注姓名和日期。
第一百七十五条 批生产记录的内容应当包括:
(一)产品名称、规格、批号 ;
(二)生产以及中间工序开始、结束的日期和时间;
(三)每一生产工序的负责人签名;
(四)生产步骤操作人员的签名;必要时,还应当有操作(如称量)复核人员的签名;
(五)每一原辅料 的批号以及实际称量的数量(包括投入的回收或返工处理产品的批号及数量);
(六)相关生产操作或活动、工艺参数 及控制范围,以及所用主要生产设备 的编号;
(七)中间控制结果的记录以及操作人员的签名;
(八)不同生产工序所得产量及必要时的物料平衡 计算;
(九)对特殊问题或异常事件的记录 , 包括对偏离工艺规程 的偏差情况的详细说明或调查报告 , 并经签字批准
社区用户@圣人有点冷:
回答:
根据“GMP 第一百六十条 应当尽可能采用生产和检验 设备自动打印的记录、图谱和曲线图等,并标明产品或样品的名称、批号和记录设备的信息,操作人应当签注姓名和日期。”和“药品记录与数据管理要求(试行) :第二十四条 对于活动的基础信息数据和通过操作、检查、核对、人工计算等行为产生的行为活动数据,应当在相关操作规程和管理制度中规定记载人员、记载时间、记载内容,以及确认与复核方法的要求。”的要求,建议采用自动打印记录,原则上,打印记录可以理解为原始数据,从数据管理和可追溯性原则来看,需要贴到批生产记录上,并且带有时间戳。
依据:
1.药品生产质量管理规范
2.药品记录与数据管理要求(试行)
社区用户@牧魂:
按照《药品记录和数据管理规范》 对原始数据 的要求,原则上对电子称打印纸,即原始记录(真实副本),需要保留的。但是实际情况看,可能不少药企并没有对备料环节进行严格管控。
当然,为了更好的落实,2021年7月23日湖南省局给出了具体的可以落实的做法:湖南省药品监督管理局关于加强药品生产记录与数据管理的通知 第三条要求:药品生产企业所有药品生产车间至少有1道关键称量 工序使用具有自动打印功能和数据可追溯的称量衡器。
如果需要附上,那么相应的时间,必须也同时具备。
关于是否必须,建议通过ERP等系统进行领料并将领料单附于批生产记录,包括退库单,以可以进行物料平衡 ,同时也可以保证其符合ALCOA+原则。
如果提问者所体现的“备料”只是领料环节的一个操作,后面有领料单,退库单。那么这个过程操作,不一定需要体现。只不过,多了操作环节,是否有SOP 规定,有无污染交叉污染 、误用等的风险。
(登录识林社区,可查看回答中涉及的法规原文链接)
作为识林知识平台的关键板块,识林社区是一个面向制药业从业者的互动交流平台,主要功能包括问答、文章、课程 。
其中,“问答”功能更是重中之重。
在这里,成百上千的识林用户提出问题,而回答问题的有其他用户,还有识林向导,识林内部团队和专家。
药品生命周期 ,是大量法规、知识、实践的结合,大家每天都会遇到大大小小的问题,但并不总能得到好的答案。
一个好的答案,不能是一句道听途说,也不仅是从“经验”出发的一句简单判断。更何况,越重要、越复杂的问题,往往并无完全正确、拿来即用的答案。
所以,优质解答
这当然很不容易,而识林向导团队本着分享和学习的精神,已经为社区带来近100份这样的优质解答。在此,识林十分感念!
接下来,我们需要的,是您的参与:
——开放 ,识林社区,目前开放给所有会员;
APP端,下载“识林”APP注册后使用,在“讨论区”提问和问答即可;PC端,复制www.shilinx.com至浏览器,点击右上角“注册”后注册登录
——解惑 ,您可以随时提问,也可以浏览全部问答;
——分享 ,识林当然也欢迎您的回答,如您也想提供优质答案,请联系我们;
——关联 :优质解答的“依据”往往包括链接,企业会员可点击跳转,普通会员可按名称检索app或监管部门网站。
识林® 版权所有,未经许可不得转载。
必读岗位及工作建议:
QA:确保所有变更符合指导原则要求,监督变更实施过程。 注册:熟悉变更分类和程序,准备和提交变更申请。 临床:参与重大变更的临床研究设计,评估变更对临床使用的影响。 文件适用范围:
文件要点总结:
变更分类明确 :根据变更对药品安全性、有效性的影响,将变更分为重大变更(A类和B类)、中等变更和微小变更。变更程序规范 :详细规定了补充申请、备案管理和年度报告等不同变更类型的申报程序。技术要求具体 :强调了变更研究技术考虑和变更申报资料的具体要求,确保变更科学合理。变更影响评估 :要求药品上市许可持有人评估变更对药品安全性、有效性、临床使用的影响。沟通交流鼓励 :鼓励与监管机构进行沟通交流,以确保变更申请的顺利进行。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位:
工作建议:
QA:确保所有文件和记录符合GMP要求,定期审核和更新文件管理系统。 生产管理:监督文件的起草、修订、审核、批准流程,确保生产活动符合文件规定。 研发:在文件起草阶段确保研发数据和结果的准确性,与生产文件保持一致。 注册:在注册文件准备过程中,确保所有文件符合监管机构的要求。 适用范围:
文件要点总结:
文件管理原则: 强调企业必须建立文件管理的操作规程,确保文件内容的正确性和一致性。质量标准: 规定物料和成品必须有经批准的现行质量标准,包括基本信息、检验方法、限度要求等。工艺规程: 要求每种药品的生产批量和包装形式都有经批准的工艺规程,且不得随意更改。批生产记录: 每批产品必须有批生产记录,记录生产过程中的关键信息,确保可追溯性。操作规程和记录: 所有关键活动都应有操作规程和记录,包括确认、验证、设备维护等。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责监督和确保药品生产过程符合M7(R1)指南要求,确保基因毒性杂质控制在可接受水平。 R&D(研发):在新药研发过程中,需评估和控制合成过程中产生的基因毒性杂质,确保药物安全性。 临床研究:在临床试验阶段,需根据M7(R1)指南评估药物中的基因毒性杂质,确保受试者安全。 注册:负责药品注册文件的准备和提交,需根据M7(R1)指南提供基因毒性杂质的评估和控制策略。 工作建议:
QA:定期审查生产流程,确保基因毒性杂质的控制措施得到有效实施,并符合M7(R1)指南要求。 R&D:在药物合成和制剂开发阶段,主动识别和评估潜在的基因毒性杂质,采取控制措施以降低风险。 临床研究:在设计临床试验方案时,考虑基因毒性杂质的风险评估,并在试验报告中包含相关数据。 注册:在药品注册文件中明确说明基因毒性杂质的评估结果和控制策略,确保符合监管要求。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):确保所有生物制品的注册申报资料符合NMPA要求。 注册:负责根据生物制品注册分类准备和提交申报资料。 研发:根据注册分类进行相应的药学、非临床和临床研究。 适用范围:
文件要点总结:
注册分类明确 :“应当”根据生物制品的特点和上市情况,明确其注册分类。申报资料要求 :强调了按照CTD格式撰写申报资料,并符合相关法规及技术指导原则。药学研究递进性 :指出药学研究在不同申报阶段是逐步完善的过程,且不同生物制品具有各自特点。临床试验数据库提交 :规定了完成临床试验后,应在CTD基础上提交临床试验数据库的具体要求。境外临床试验数据要求 :特别指出境外申请人在境内开展临床试验需满足的特定数据要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位建议:
QA:确保记录与数据管理符合法规要求,监督记录的准确性和完整性。 生产:遵循记录管理规程,保证生产过程的记录真实、准确。 研发:确保研发过程中数据的采集、处理和存储符合规定。 注册:在药品注册过程中,确保提交的记录与数据符合监管要求。 适用范围说明:
文件要点总结:
记录与数据定义: 明确了数据和记录的定义,包括其类型和形式。记录类型与管理: 规定了记录的类型、管理措施、以及电子记录与纸质记录的同等功能要求。电子记录系统要求: 详述了电子记录系统的设施配置、功能要求和用户权限管理。数据管理规范: 包括基础信息数据、行为活动数据、计量器具数据和电子数据的管理措施。记录与数据的生命周期管理: 从记录的创建、更改、保存到销毁的全过程管理要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位解读:
QA(质量保证) :必读。应确保所有生产活动符合GMP要求,监控生产过程,保证产品质量。注册 :必读。需熟悉GMP规范,以确保注册文件和申报材料的合规性。生产 :必读。应根据GMP要求组织生产活动,确保生产过程的规范性。研发 :参考。在产品开发阶段考虑GMP要求,为后续生产打下基础。临床 :参考。了解GMP对临床试验用药品的特殊要求,确保临床试验的合规性。适用范围说明:
文件要点总结:
原料血浆管理 :强调原料血浆的质量和来源合法性,以及生产过程中病毒去除和/或灭活的严格控制。人员资质要求 :明确企业负责人、生产管理负责人、质量管理负责人和质量受权人的资质和经验要求。生产设施和设备 :规定血液制品生产厂房、实验室的独立性和专用性,以及防止交叉污染的措施。原料血浆检验与追溯 :要求企业对原料血浆进行严格检验,建立追溯系统,确保可追溯性。生产和质量控制 :强调生产过程中的温度验证、体外诊断试剂管理、病毒污染和安全风险评估,以及信息化记录的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。