首页
>
资讯
>
三方共建药典各论:FDA、USP 与 AAM 会商药典新战略
出自识林
三方共建药典各论:FDA、USP 与 AAM 会商药典新战略
2026-02-05
{{#video:vf807f172-8de0-4fb0-a606-cf3500d09bd0}}
2025年12月11日,FDA、美国可及药品协会(AAM)与美国药典委员会(USP)联合举办题为"质量与监管可预测性:塑造USP标准"(Quality and Regulatory Predictability: Shaping USP Standards) 的网络研讨会。三方代表从监管、标准制定与产业实践角度,共同探讨了如何通过深化协作、优化标准制定流程,为制药行业构建更可预测、更高效的监管环境。
FDA与USP的协作与困境
FDA通过"政府联络员项目"(Government Liaison Program)深度参与USP标准制定,在2020-2025周期内有超过130名CDER工作人员在USP专家委员会中任职。这种协作旨在确保USP标准与FDA监管思路保持一致,为行业提供清晰的合规指引。
研讨会上FDA CDER-COSS(Compendial Operations and Standards Staff)主任Pallavi Nithyanandan博士坦承当前标准制定中面临的最大挑战:FDA无法向USP披露申请中的企业特异性信息(如杂质谱 、接受标准 ),因为这些数据被视为企业机密,存在于新药申请(NDA) 或药物主文件(DMF) 中;而USP缺乏这些数据,则难以制定或更新符合现代质量要求的各论。这种信息不对称导致"先有鸡还是先有蛋"的困境——FDA不能批准不符合USP各论的申请,而USP又因申请尚未获批而无法修订标准。
PMP流程共建药典各论:破解"信息共享"困局
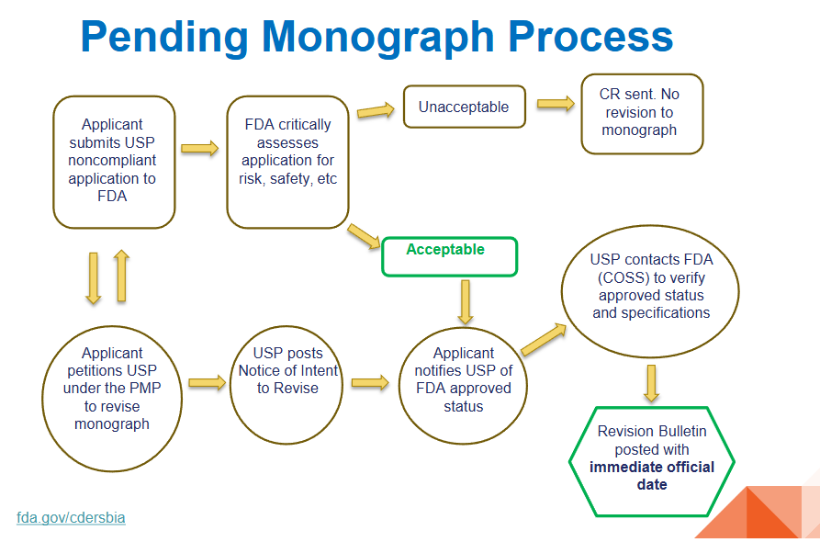
为解决这一瓶颈,Nithyanandan博士重点介绍了"待决各论流程"(Pending Monograph Process, PMP)。该流程允许申请人 在FDA审评期间主动发起专论修订请求,一旦FDA认为申请可接受,USP即可发布"修订意向通知",并在FDA正式批准后迅速将标准修订为官方状态,赋予立即生效日期。根据Nithyanandan博士展示的流程图:
PMP的具体运作步骤如下:
第一步:申请人提交非合规申请
第二步:FDA批判性评估
若评估为"不可接受":发出完整回复函(CR),不触发USP专论修订。
第三步:申请人向USP申请修订(PMP请愿)
申请人在获知FDA"可接受"结论后,正式向USP提交修订专论的请愿(petition)。
USP发布"修订意向通知"(Notice of Intent to Revive),公示拟议的专论变更 。
第四步:通知与验证
USP联系FDA(通过COSS)验证批准状态及具体规格参数。
第五步:发布修订公告(Revision Bulletin)
经确认后,USP发布修订公告(Revision Bulletin),赋予该专论修订立即生效的官方日期(immediate official date)。
Nithyanandan博士指出,这一机制有效加速了505(b)(2)申请 和仿制药 (ANDA)的批准进程,避免了因标准不符导致的市场准入延迟。
监管呼吁企业参与,共建透明、及时的信息体系
USP专论开发采用全生命周期 方法,遵循五个明确阶段:启动、开发中、药典论坛(Pharmacopeial Forum,PF)公示、专家委员会(EC)审查、正式发布。
AAM科学与监管事务高级总监Scott Kuzner详细描绘了药典流程与利益相关方输入的双向互动机制,并建议企业在"开发中"阶段即与USP接触,以获得可控的数据收集与内部审查,主动塑造标准;将参与从被动的"PF评论"转向主动的"早期介入",在监管生命周期早期共享信息可避免未来合规差距,增加监管确定性。
为确保"监管可预测性",产业界必须掌握USP的信息发布渠道与时间节点。USP行业参与总监Matthew Vankoski特别提醒,企业应密切关注USP-NF在线平台的"New and Changed"更新和"Annotated List",利用这些免费资源建立内部监测流程,确保在90天评论期内及时提交数据与意见。FDA也在批准函中新增了明确条款,要求申请人 与USP直接合作修订官方各论,以确保获批产品持续符合药典标准。
FDA、USP与产业界的三方协作,特别是通过PMP等创新机制破解信息不对称,是提升监管可预测性的关键路径。正如三位发言人共同强调的——无论制造商是谁,所有患者都有权获得质量合格的药品;而企业通过早期参与、数据共享与积极倡导,不仅能保护自身合规利益,更能塑造一个更稳健、更透明的全球药品质量生态系统。
识林-白蜡
识林® 版权所有,未经许可不得转载
岗位必读建议:
QA(质量保证):关注药典通告更新和药典论坛更新,确保公司产品符合最新的USP标准。 R&D(研发):在新药开发过程中,参考USP-NF介绍和USP与FDA相关内容,确保研发流程和产品质量符合规定。 Production(生产):依据USP-NF的具体要求,调整生产流程和质量控制标准。 Regulatory Affairs(注册事务):密切关注修订公告和中期修订声明,及时更新注册文件和策略。 文件适用范围:
本文适用于所有在美国市场销售的化学药品和生物制品,包括创新药和仿制药。发布机构为美国药典(USP),企业类别包括Biotech、大型药企、跨国药企以及CRO和CDMO等。
要点总结:
药典通告更新 :强调了药典通告的及时性,包括一般公告、修订意向通知和出版物更正。药典论坛 :提供了公众评议的平台,包括提议的中期修订声明和常规修订。修订公告 :作为药典标准最快的修订途径,解决紧急问题,如病人安全性和纠正重要错误。中期修订声明 :加速修订的形式之一,解决重要性次于修订公告的议题。勘误 :纠正印刷错误,不具有广泛影响。以上仅为部分要点,请阅读原文,深入理解监管要求。