|
首页
>
资讯
>
国内药政每周导读:国家级政策深化药品监管改革,辅料包材 GMP 落地,肽类、抗体、CAR-T 和胰岛素等多个临床指南定稿
出自识林
国内药政每周导读:国家级政策深化药品监管改革,辅料包材 GMP 落地,肽类、抗体、CAR-T 和胰岛素等多个临床指南定稿
2025-01-06
祝识林用户2025新年快乐!
【早期研发与临床】
12.31,【CDE】关于发布《评价胰岛素类药物药代和药效动力学的正葡萄糖钳夹试验指导原则》的通告(2024年第56号)
本文曾于2024年6月征求意见。
随着胰岛素类药物的广泛应用,近年来新型胰岛素制剂逐渐成为研发的热点。不同胰岛素类药物的区别主要是药代动力学(Pharmacokinetics,PK)和药效动力学(Pharmacodynamics,PD)差异。正葡萄糖钳夹试验由于能很好地排除内源性胰岛素的影响,客观反映外源性胰岛素类药物的 PK 和 PD 特点,目前在胰岛素类药物的临床评价中成为国际公认的可靠方法。
本指导原则主要涉及采用正葡萄糖钳夹试验评价胰岛素类药物的 PK 和 PD 特征,适用于胰岛素类药物的临床评价,对于每周一次的长效胰岛素类药物的 PK 和 PD 研究设计,可与药审中心进行沟通交流。
12.31,【CDE】关于发布《腺相关病毒载体基因治疗产品非临床研究技术指导原则》的通告(2024年第57号)
本文曾于2024年7月征求意见。
本指导原则所指 AAV 载体基因治疗产品(以下简称为“AAV 载体产品”)是以 AAV 载体携带含目的基因的基因表达元件,通过目的基因在体内发生转录、翻译,获得目的蛋白并在体内长期表达来发挥治疗作用。该类产品的潜在风险分析时需考虑终产品、所采用的 AAV 载体、递送的目的基因等,还需关注体内长期存续和/或表达带来的长期安全性风险。
对于携带基因编辑工具的 AAV 载体产品,其作用机制涉及对人体细胞基因组的编辑操作,现阶段对此类产品的认知有限,因此本指导原则不适用于此类产品。
12.31,【CDE】关于发布《嵌合抗原受体 T 细胞治疗血液淋巴系统恶性肿瘤临床试验技术指导原则(试行)》的通告(2024年第58号)
本文曾于2024年8月征求意见。
本指导原则主要适用于利用患者自体细胞制备的CAR-T细胞产品,也可适用于利用异体供者细胞制备的异体或通用型 CAR-T 细胞产品,或者其他类型的基因修饰淋巴细胞产品如CAR 自然杀伤细胞(Chimeric antigen receptor -Natural Killer cells,CAR-NK)或T细胞受体工程 T细胞(Engineered T cell receptor-T cell, TCR-T)等。
本指导原则适用的血液淋巴系统恶性肿瘤主要包括 B-ALL、B-NHL和 MM等肿瘤类型。不同产品类型或不同肿瘤分类人群开展临床试验的考虑可能存在差异,关于其他类型基因修饰淋巴细胞或肿瘤分类的具体考虑因素,建议根据具体情况与药审中心进行沟通。
12.31,【CDE】关于发布《预防用猴痘疫苗临床试验技术指导原则(试行)》的通告(2024年第59号)
本文曾于2024年8月征求意见。
猴痘病毒主要经黏膜和破损皮肤侵入人体,主要通过直接接触患者的病变皮肤或黏膜传播,接触感染动物的呼吸道分泌物、病变渗出物、血液及其它体液,或被感染动物咬伤、抓伤传播,亦可通过接触被病毒污染的物品、长时间近距离吸入感染者呼吸道飞沫传播。
本指导原则是根据预防用疫苗相关临床研究技术指导原则,同时结合境内外猴痘疫苗的研究数据和研发现状,基于现有科学认知水平形成的共识,用于指导猴痘疫苗的研发和临床评价。
12.31,【CDE】关于发布《脑膜炎球菌疫苗临床试验技术指导原则(试行)》的通告(2024年第60号)
本文曾于2023年12月征求意见。
脑膜炎球菌的荚膜多糖(capsule polysaccharides,CPS )是主要的致病物质,可诱导机体产生血清杀菌抗体,激活补体介导的裂解和增强调理吞噬作用等来提供保护作用,针对A、C、W和Y群等血清群脑膜炎球菌的疫苗主要分为两类,分别为多糖疫苗(meningococcal polysaccharide vaccine, MPSV, 其抗原为 CPS)和多糖结合疫苗(meningococcal polysaccharide conjugate vaccine, MPCV, 其抗原为 CPS-蛋白载体)。
本指导原则旨在为采用CPS 或CPS-蛋白载体为有效抗原成份的脑膜炎球菌疫苗临床试验设计及评价提供建议。
1.2,【CDE】关于发布《肽类药物临床药理学研究技术指导原则》和《抗体类药物临床药理学研究技术指导原则》的通告(2024年第55号)
两文分别于2024年6月和7月征求意见。
两类药物都是目前创新的热点,前者代表是GLP-1靶点药物司美格鲁肽,后者则包括单抗、双抗,不过ADC不在其讨论范围内。
点击“花脸稿”可以看到抗体文的改动较多,肽类文较少,但这些修订背后都有监管与风险考量,建议识林会员仔细阅读花脸稿。
【CMC与仿制药】
1.2,【CDE】关于公开征求《化学仿制药参比制剂目录(第九十批)》(征求意见稿)意见的通知
征求意见截至1月15日。
截至目前,NMPA已发布参比制剂目录87批,CDE已公示90批。
识林用户可登录“中国参比制剂库”查阅。
【注册审评与变更】
12.31,【CDE】关于公开征求《简化港澳已上市传统口服中成药内地上市注册审批申报资料及技术要求(征求意见稿)》意见的通知
征求意见截至1月14日。
本文件主要内容包括“上市注册”、“上市后变更”和“再注册”三个部分。第一部分“上市注册”明确了“港澳已上市传统口服中成药”在内地申请上市注册所需的行政文件、药学资料和上市支持性资料、说明书及包装标签等相关要求。第二部分和第三部分明确了上市后变更和再注册的相关要求。
考虑到适用品种为在香港、澳门特区已上市的传统口服中成药,其原在香港、澳门特区上市注册时提交的试验研究资料可作为相应的申报资料。各项资料要求都做了相当的减免。
1.3,【CDE】常见一般性技术问题解答 - 新增2个问答
- 已在境内上市的境外生产原研药品转移至境内原研企业持有后,如何遴选参比制剂?
截至2025-01-03,官网共发布一般性技术问题解答237条(其中有25条重复,实际共计210个)
CDE 化学仿制药共性问题、CDE 受理共性问题可点击查看。
【生产质量与检查】
12.30,【CFDI】关于公开征求《粉液双室袋产品检查指南(征求意见稿)》意见的通知
征求意见截至1月15日。
本指南作为粉液双室袋产品注册现场核查及药品生产质量管理规范符合性检查的技术指导文件,基于粉液双室袋产品目前的生产技术条件、现有法规框架和粉液双室袋产品特点,分析可能影响产品质量的风险,阐述粉液双室袋产品在注册研制和生产现场核查与药品生产质量管理规范符合性检查中不同于常规制剂的检查重点。
本指南仅针对弱焊结构的粉液双室袋产品。
12.31,【上海】关于印发《上海市药品监管部门规范涉企行政检查实施方案》的通知
这篇文的背景也许是上周国务院常务会议通过的《关于严格规范涉企行政检查的意见》。
上海药监的做法包括:
检查对象分级:市药品监管部门按照“产品+体系+信用”建立评价模型并迭代升级,对检查对象进行综合评价,并划分为A、B、C、D四个等级进行管理,制订升降级规则...纳入“无感监管”的,除上级部署、收到违法线索和法律法规规章有明确规定的外,原则上不主动开展现场检查。
无码不检查。有计划行政检查或触发式行政检查需启动现场检查的,均需应用“检查码”。行政执法人员应在现场检查前,报请本部门负责人同意后,通过上海市药械化行政检查系统填写检查相关信息后申请“检查码”,并按相关要求向检查对象推送“检查码”。
不过,以下检查行为暂不纳入本次规范行政检查的范围:(1)以告知承诺等方式获得行政许可的,对告知承诺事项的核查行为;(2)行政处罚立案后的询问检查、整改复查等行为;(3)对药品、医疗器械和化妆品等质量安全的抽检行为;(4)对无照经营者的检查行为;(5)按保密规定开展的涉密检查行为;(6)对外省市经营主体在本市管辖范围内开展的检查行为。
1.2,【NMPA】关于发布《药品生产质量管理规范(2010年修订)》药用辅料附录、药包材附录的公告(2025年第1号)
国家局新年第一号文件,是两篇新GMP附录落地。
查看版本历史可知,辅料GMP早在2006年发布,2024年7月征求意见。包材GMP曾于2022和2024两次征求意见。
辅料包材供应商当然需要仔细阅读并执行,想来会有大量质量体系文件要升版。
但不仅如此,用“页面内搜索”,可见两文中均有多处出现“药品生产企业”(包材GMP中提及,与留样有关)和“药品上市许可持有人”(辅料和包材GMP均提及),这也意味着作为客户的药企,尤其是质量部门,也应关注这两篇GMP附录,作为供应商管理的依据。
【监管政策综合】
12.30,【上海】关于公布全国药品集中采购(GY-YD2024-2)中选结果的通知
10批集采落地。
有媒体分析,与拟中选结果相比,仅有的变化是枸橼酸坦度螺酮片的最终中选企业减少1家。
回顾第十批国采,申报企业439家,涉及产品778个,从最终的中选结果来看,共有380多个产品中选,涉及230多家企业,其中有110余家是B证企业,占比近1/3。
据相关统计数据,第十批国采的产品拟中选率约49.5%,是历年国采中最低的一批;企业拟中选率约53.5%,同样是自“4+7”扩围以来最低的一批。
针对第十批国采,12月26日,国家医保局召开医药集中带量采购座谈会。围绕带量采购,座谈会重点回应了企业在加强成本控制、保障中选产品质量和供应方面采取的措施。
对于集采中选产品,国家药监局药品监管司有关工作负责人表示,药监部门对中选药品实行生产企业检查和中选品种抽检两个100%全覆盖,确保“降价不降质”。
1.3,【国务院】关于全面深化药品医疗器械监管改革促进医药产业高质量发展的意见
年初业界就迎来国家级政策,是对医药行业发展的顶层设计。与2015年的44号文,2017年的42号文属于一个层次,预期将对整个医药产业带来极其深远的影响。
文中的一些政策已经有文件发布并实施,但也有曾经提及但未能落地的举措,下面列举一些具体明确的条文,并检索部分相关文件,供识林会员参考:
- 对符合条件的罕见病用创新药和医疗器械减免临床试验。
- 将罕见病用药品注册检验批次由3批减为1批,每批次用量从全项检验用量的3倍减为2倍。基于产品风险统筹安排进口罕见病用药品注册核查与上市后检查,缩短境外核查等待时限。
- 将仿制药质量和疗效一致性评价逐步向滴眼剂、贴剂、喷雾剂等剂型拓展。
【新药批准和报产】
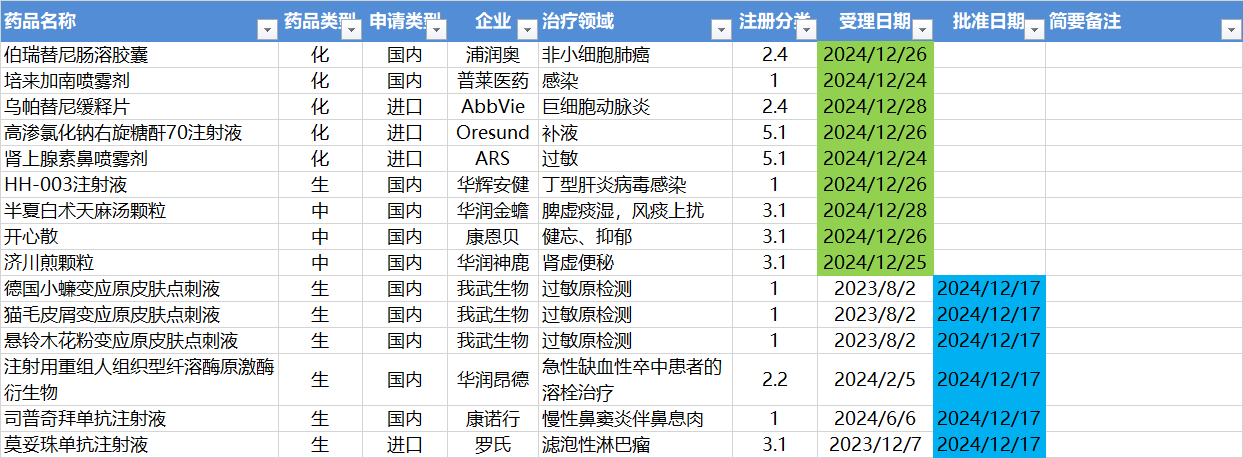
12.30-1.5,NMPA发布8个新药批准,CDE受理9个NDA
注:仅列出新药(包括改良型新药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林®版权所有,未经许可不得转载。
|