|
首页
>
资讯
>
有关FDA优先审评券的五个关键更新
出自识林
2015-09-04 FDA law blog
这篇文章中,我们着重关注与FDA热带疾病和罕见儿科疾病(“儿科”)相关的优先审评券(PRV)项目的5个关键更新:
1. 联合治疗公司(“联合治疗”)以3.5亿美元的价格出售其儿科PRV;
2. FDA在热带疾病资格名单中增加美洲锥虫病和脑囊虫病;
3. 赛诺菲-安万特(“赛诺菲”)成功兑现一张儿科PRV,击败安进,成功上市,成为在美国首个获批的PCSK9抑制剂降脂药;
4. 参议院发布更新版《推进希望法案》;
5. 3个杜氏肌营养不良症(DMD)申请人获得罕见儿科疾病认定。
PRV价格持续攀升
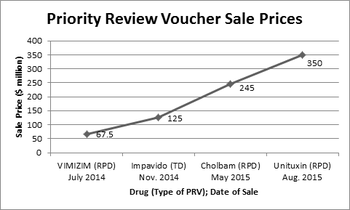
8月19日,联合治疗宣布将其拥有的罕见儿科PRV以3.5亿美元的价格出售给艾伯维公司。如下图所示,PRV的价值持续攀升。有关FDA就Unituxin的批准向联合治疗公司发放儿科券的讨论请见此处。
FDA在热带疾病资格名单中增加美洲锥虫病和脑囊虫病
8月20日,FDA公布最终裁定,将两个新疾病:美洲锥虫病和脑囊虫病,添加进热带疾病资格名单中。回顾一下,为了让一个药品有资格获得PRV,必须满足4个条件:
1. 申请必须是一个已列出的热带疾病;
2. 申请必须以505(b)(1)NDA或505(b)(2)申请提交;
3. 所申请的药品,必须不含有先前获批的活性基团;
4. 申请必须符合FDA政策下的6个月优先审评。
因此,符合上述条件的治疗美洲锥虫病和脑囊虫病的药品申请人有资格享受热带疾病RPV。
FD&C法案524(a)(3)允许FDA覆盖“任何其它在发达国家没有显著市场,而严重影响到贫困和边缘化人群的传染病。”在其最终裁定中,FDA重申其承诺,在“对候选疾病科学认知、定性评估”的基础上对热带疾病认定使用“灵活方式”。FDA还宣布建立持续开放的公共卷宗,接收未来对于热带疾病认定的建议。
最终裁定阐述了FDA对FD&C法案524(a)(3)(R)关键要素的解释:
“发达国家” – 按照裁定,“FDA将使用一个国家在世界银行‘低收入国家’的出现……作为证据,该国家不应被认为是以‘热带疾病’确认为目的的‘发达国家’。”
“没有显著市场” – FDA指出,提供“没有显著市场”严格定义的困难,而是提出了确认发达国家是否存在“显著市场”的下列考虑因素:(1)疾病在发达国家的发生率;以及(2)对于热带疾病药物相当大的间接市场的存在(例如,政府,包括军队)将构成对药物研发的经济激励。
“严重影响到贫困和边缘化人群” – 与“没有显著市场”的说法一样,FDA抛弃单一定义,支持需衡量4个因素。这些因素是:(1)归因为发展中国家疾病的全球伤残调整寿命年数的比例;(2)在被发现国家最贫困人口中的疾病相对负担;(3)在婴儿、儿童和其他边缘群体中的疾病相对负担;和(4)WHO是否将其作为被忽视的热带疾病。
应用这些标准,2014年12月,FDA行使法律授予的权利,将Ebola添加到FDA优先审评券计划法案,并将美洲锥虫病和脑囊虫病添加进热带疾病券计划的资格名单中。
额外收获:2015年8月24日,中大西洋生物治疗公司提交了一份公民请愿,请求FDA将狂犬病病毒添加进热带疾病资格名单中。值得注意的是,狂犬病像囊虫病/绦虫病一样,在WHO被忽视热带疾病名单中。
赛诺菲成功使用其PRV
8月19日,FDA宣布,赛诺菲-安万特已经为Praluent(alirocumab)兑现其儿科PRV。随着7月24日的批准,Praluent成为首个在美国批准的PCSK9降脂药。PCSK9抑制剂类药物有望成为重磅炸弹,每年销售额预计将在数十亿美元。
这个故事有趣的地方是,安进的Repatha击败赛诺菲的Praluent在欧洲上市;但,表面上看,通过其罕见儿科疾病优先审评券的兑现,赛诺菲得以在安进之前进入美国市场。Praluent批准比Repatha的2015年8月27日PDUFA目标日期和批准提早1个月。
比起后来者,首个进入市场的药物通常享有可观的收益。以美国支付方为例,他们会很快覆盖新药,首个进入市场的产品往往具有显著的营销优势和销售力。赛诺菲为其PCSK9降脂药投入6.75千万美元用于加速进入美国市场的举措将受到密切关注。毕竟,这将是证明企业是否能够支付优先审评券高昂价格的领头羊。如果赛诺菲一切顺利的话,其后PRV的价格和需求将高涨。
推进希望法案更新
2015年3月,国会议员G.K. Butterfield在众议院提出推进希望法案(Advancing Hope Act,H.R.1537)。该法案提出使儿科券计划永久化。法案还提出改变热带疾病券计划。在当时,这种变化不仅显得格格不入,还可能会适得其反。详细讨论请见此处。
2015年7月28日,推进希望法案(S.1878)引进参议院。参议院版本的提案废弃了对热带疾病优先审评券计划的修订。相反,参议院版本提出儿科券计划永久化,并认定儿科癌症和镰状细胞贫血症为罕见儿科疾病。
杜氏肌营养不良症(DMD)获得罕见儿科疾病认定
8月19日,FDA授予BioMarin公司、Sarepta公司和Santhera公司研发的杜氏肌营养不良症产品罕见儿科疾病认定。随着罕见儿科疾病的认定,每家公司均将有可能在其产品批准之上获得儿科券。
参考资料:
原文地址
识林TMwww.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
适用岗位: - 注册(RA):必读。需了解FDA优先审评券计划的更新,特别是针对埃博拉病毒治疗药物的审评政策。
- 研发(R&D):必读。应关注埃博拉病毒相关药物研发的激励政策,评估项目优先级。
- 临床(Clin):必读。了解埃博拉病毒治疗药物的审评时间缩短,对临床试验设计的影响。
工作建议: - RA:评估现有和未来项目是否符合优先审评券计划条件,制定注册策略。
- R&D:优先考虑埃博拉病毒治疗药物的研发,利用政策激励加速项目进展。
- Clin:根据审评时间缩短,调整临床试验设计和时间规划,加快药物上市进程。
适用范围:
本文适用于针对埃博拉病毒治疗药物的化学药和生物制品,适用于创新药和生物类似药,由美国FDA发布,适用于Biotech和大型药企。 要点总结: - 埃博拉病毒纳入优先审评券计划:明确将埃博拉病毒治疗药物纳入FDA优先审评券计划,以鼓励相关药物的研发。
- 无限制转让次数:取消了优先审评券在首次使用前转让次数的限制,增加了政策的灵活性。
- 审评时间缩短:将审评券的审评时间从365天缩短至90天,加快了埃博拉病毒治疗药物的审评进程。
- 法规修订:修订了联邦食品、药品和化妆品法案第524节,正式将埃博拉病毒治疗药物纳入优先审评券计划。
- 政策生效:该法案于2014年12月16日生效,为埃博拉病毒治疗药物的研发和审评提供了明确的政策支持。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |

