|
首页
>
资讯
>
监管科学大会记要:生物药(单抗)企业的FDA检查缺陷研究
出自识林
监管科学大会记要:生物药(单抗)企业的FDA检查缺陷研究
2023-04-17
2023年4月1-2日第六届中国药品监管科学大会在北京召开,会议共有 14 个分论坛,覆盖了“两品一械”相关领域,药监机构、科学界、医疗机构、制药业以及相关协会代表 1800 余人参加了会议。
在生物制品监管专题研讨会中,识林兼北京大学知识工程与监管科学实验室顾问王国旭研究员做了题为“生物药(单抗)企业的FDA检查缺陷研究”报告,本文简要概述报告要点。
研究背景
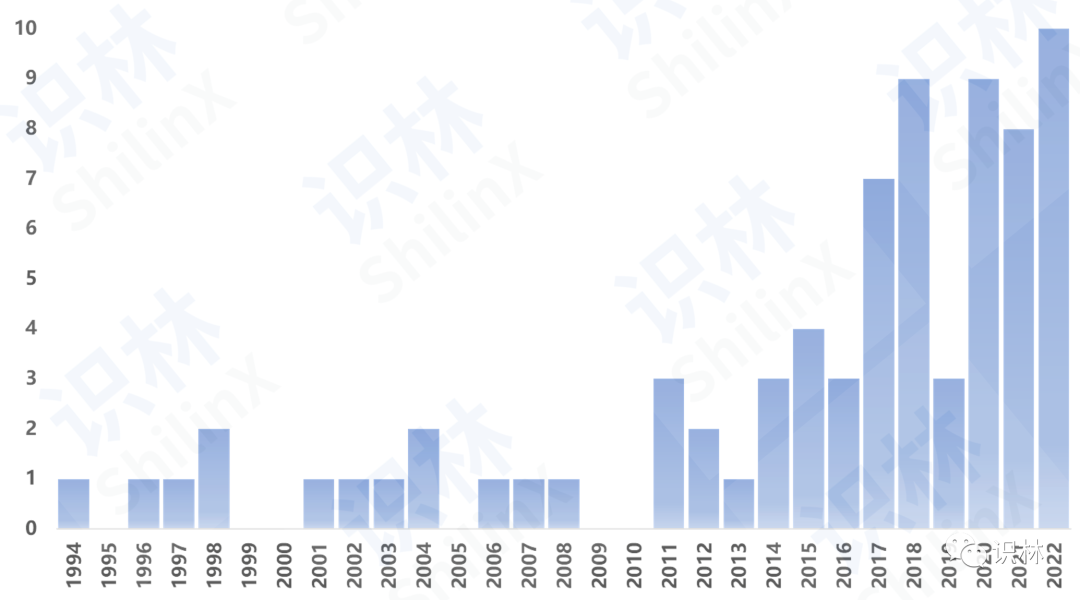
近年来,随着首次获得FDA单抗药物批准的企业数量不断增加,单抗企业越来越呈现小散多的趋势。
研究方法和结果
FDA单抗产品检查类型主要为两类,第一类为许可前检查(PLI),旨在评价一份已提交待审批的生物制品许可申请(BLA);第二类为GMP监督检查,通常每两年一次,基于风险和关键任务,目的主要是核实企业是否符合CGMP要求。
FDA对检查缺陷的记录主要是通过483和警告信体现。识林483和警告信数据库收录了2008年至今15年的检查数据,其中483数据库现有近8万5千条检查信息和约1.5万封483原文,警告信数据库现有570封中译警告信。本研究的样本数据就是基于数据库中单抗企业相关的检查结果,样本范围包括48个单抗生产设施的286次检查。
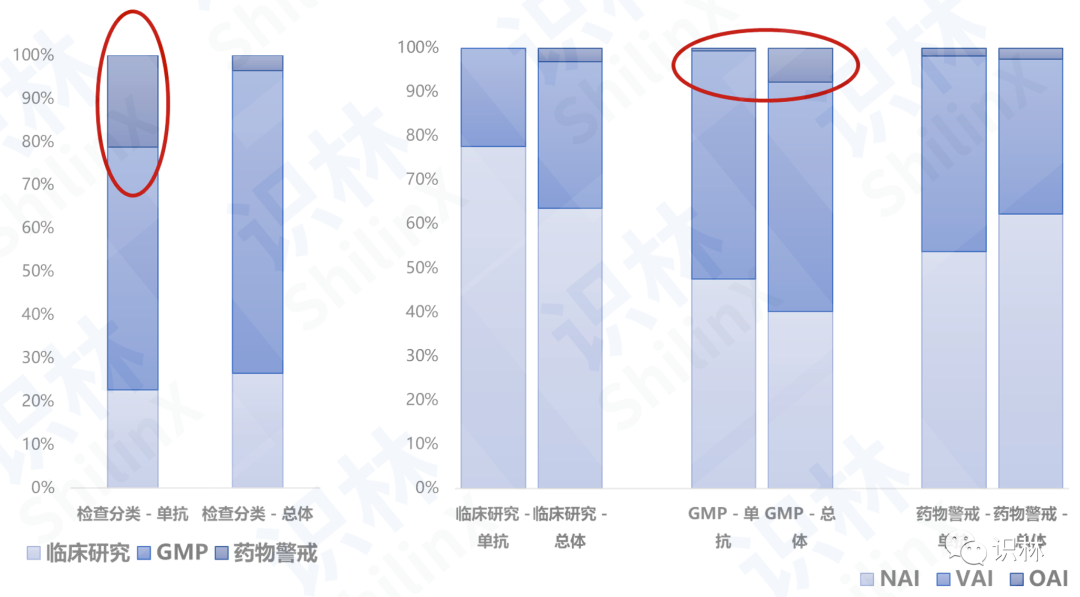
从检查分类来看,相比于总体,单抗企业药物警戒检查所占比例更多,GMP检查更少;从检查结果来看,相比于总体,单抗企业GMP检查中OAI所占比例更少,NAI和VAI所占比例更多。
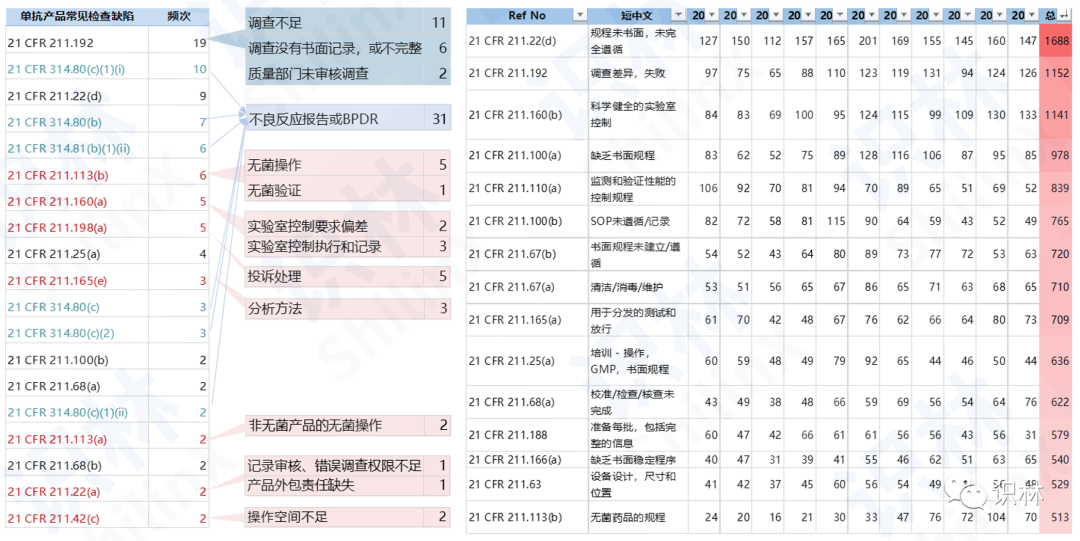
从缺陷类别分析,单抗产品常见的检查缺陷包括对偏差的调查和记录不完善,不良反应或生物制品偏差报告不足,以及无菌操作和实验室控制相关缺陷。
FDA在2021年发布了蛋白原液生产企业监督检查指南,旨在从质量体系的六大系统指导检查员蛋白原液监督检查的考虑因素,以及一些关键缺陷的示例。本研究针对指南中列举的六大系统关键缺陷,都研究了具体的实例,并逐一进行了483中相关缺陷项和具体细节的分析。
完整案例:单抗企业检查缺陷分析和回复策略思考
报告介绍了两个完整案例。第一个为检查回复案例,FDA在2017年对Celltrion进行了GMP常规检查,并签发了59页长的检查报告(EIR),以及包含12条缺陷的483。Celltrion几周后给出了142页的483回复,逐条回应检查缺陷,并伴随着大量的分析图表,但在2018年仍然收到了警告信。分析原因可以发现,483中列举的缺陷,包含数据可靠性、无菌操作、质量体系等系统性问题,而企业的回复则是凌乱分散、就事论事、堆砌数据、浅尝辄止。此外,可以看到该企业警告信中所列举的缺陷项无一不在483中已经指出。因此本研究也涉及对483和警告信缺陷项的关联分析,即哪些缺陷项在483中出现后也较大可能会在警告信中出现。
报告中的第二个案例为Biocon Sdn 马来西亚工厂连续五年 483 解析,该案例分析了工厂2018年-2022年连续5年的检查缺陷,以及历年检查缺陷的重复情况,其中出现频率较高的缺陷项主要为:没有充分遵循书面规程、偏差调查不彻底、设备清洁和维护不足、未建立和遵守无菌产品免受微生物污染的规程。该案例的具体分析,可登录识林查看。
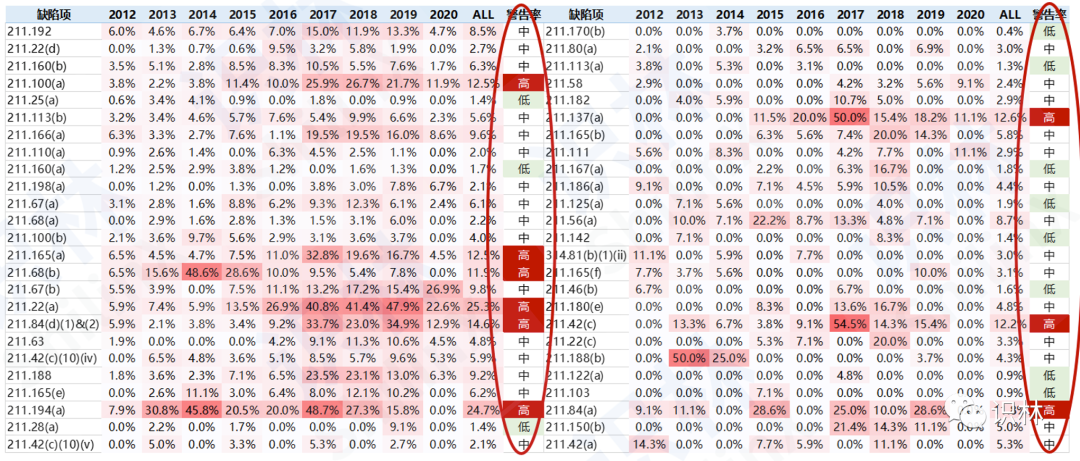
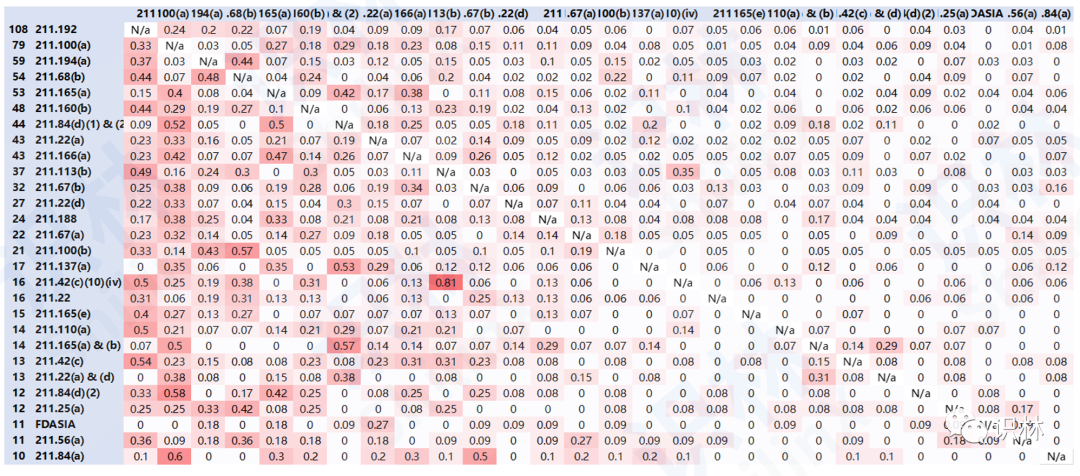
报告最后总结了两条经验和3个分析维度。经验1:缺陷是相互关联的,切忌头疼医头,脚疼医脚。通过图8的分析可以看到,FDA所引用的缺陷项是有一定相关性的,即有些缺陷项经常会一同出现,这也是FDA对企业回复的期待:除了对所指明的缺陷项本身,企业是否还会考虑上升到背后的根本原因,以及与此相关的缺陷项整改。
经验2:483、警告信和缺陷回复是实用的大数据。这里引用了识林案例:警告信中关于培养基模拟灌装的合规风险详解 -- 如何使用警告信数据库理清合规红线。通过运用警告信数据库分析培养基模拟灌装的合规风险,告诉听众如何运用检查大数据来分析某一具体类别或操作的合规风险。
此外,通过以上案例的分析,运用483和警告信等检查大数据,可以从3个维度进行检查缺陷分析:1.纵向维度,分析某个具体企业历年完整的检查案例,得出对于该企业检查缺陷的分析和整改回复的思考;2.横向维度,通过分析某一类企业甚至某个具体操作的检查结果,得出对于该类企业或具体操作的合规风险;3.点对点维度,如分析FDA某位检查员历史检查结果,得出该检查员常见的检查关注点,或者针对某个国际事件(如新冠),分析FDA检查方式和检查关注点的变化。
识林-雪杉
识林®版权所有,未经许可不得转载
在解读FDA_CPGM_7356.002M_Surveillance_Inspections_of_Protein_Drug_Substance_Manufacturers_202108这份文件时,以下是对该文件的解读和建议: 适用岗位: - 本文件是针对蛋白类药物原料生产企业的监管检查指南,因此对于以下岗位是必读的:
- QA(Quality Assurance):确保生产过程符合CGMP要求。
- 生产部门:涉及蛋白类药物原料的具体生产操作。
- 研发部门:涉及蛋白类药物原料的研发和工艺开发。
- 质量控制实验室(QC Laboratory):负责原料和产品的检验工作。
工作建议: - QA应制定或更新内部审计计划,确保涵盖本指南中提到的所有关键点。
- 生产部门应根据本指南调整生产操作,确保符合最新的监管要求。
- 研发部门应在药物开发阶段考虑本指南的要求,确保可生产性。
- QC实验室应根据本指南更新或验证检验方法,确保检验结果的准确性和合规性。
文件适用范围:
本文适用于美国FDA监管下的蛋白类药物原料生产企业的监管检查,包括但不限于重组和非重组蛋白原料,不包括疫苗、中药等其他类型的药品。 要点总结: - 监管检查范围限定:本指南仅限于对蛋白类药物原料生产企业的监管检查,与CDER和ORA的协议一致。
- 数据报告要求:特别指出了蛋白类药物原料制造CGMP检查的附加报告要求。
- 不良事件报告:检查团队在发现不良事件报告不足时,应及时通知CDER的OQS。
- 检查报告完成:ORA部门需在检查结束后45天内完成EIR的电子上传。
- 潜在OAI行动:ORA部门应及时更新Panorama和eNSpect,当潜在的OAI行动被指示时。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。
- 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。
- 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。
适用范围:
本文适用于涉及化学药、生物制品、疫苗和中药等药品类型的企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于不同规模的企业,如Biotech、大型药企、跨国药企、CRO和CDMO等,由相关药品监管机构发布。 文件要点总结: - 质量管理体系概述:明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。
- 产品质量实现要素:涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。
- 质量保证要素:包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。
- 质量风险管理:介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。
- 质量管理系统文件:规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |