研究者表示,EMA 与 FDA 之间在上市批准初步决定中的一致性高达 91%,而在重新提交或重新审查的申请审评方面的一致性达到 98%。总体而言,84%(90/107)的申请在首次提交到两家机构时获得批准,其中 EMA 的首轮批准率(92%)高于 FDA(85%)。研究指出,“FDA 的初始不批准决定(FDA 13%,EMA 3%)反映了多种因素,这些因素因不同机构对疗效的结论不同而有所不同,这并不奇怪,因为独立评价的判断会存在一些差异。”
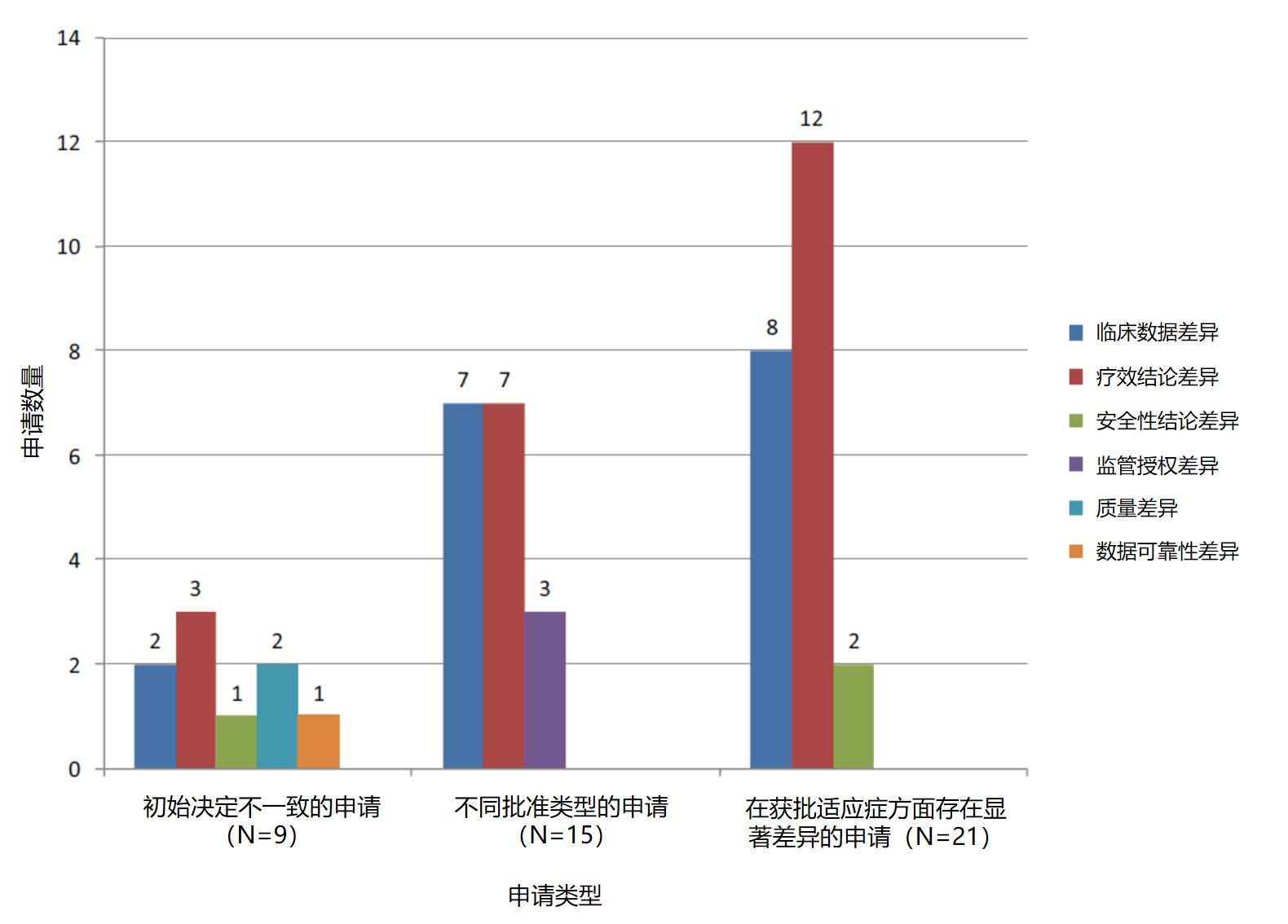
导致差异的第二个最常见原因是为支持申请而提交的临床数据的差异。EMA 在一份声明中解释指出,在临床数据中观察到的一些差异是由于递交时间的不同而导致的 — 更多的申请在提交到 EMA 之前首先提交到 FDA。与 FDA 相比,EMA“经常审评包含更多临床试验的申请,或者尤其是对抗肿瘤药来说,包含来自同一临床试验的更成熟数据的申请。在这些情况下,EMA 比 FDA 更有可能给予标准批准、更广泛的适应症或药品作为一线治疗来使用。”导致初始批准决定不一致的其它原因还包括有关支持安全性的证据强度的不同结论,其中一个机构对申请中数据可靠性或申请人是否遵守 GMP 的担忧。
研究作者表示,虽然之前有过对两家机构监管批准的比较,但都各有限制,他们的最新研究是迄今为止最全面的研究,标志着两家机构首次努力比较他们与上市申请相关的决策以及任何差异产生的原因。他们大胆地提出,FDA 和 EMA 决策的高度一致“表明正在开展的监管科学合作的努力可能有助于该领域的全球协调,包括应用于药物开发的临床药理学和转化科学。”
研究审查的 107 件上市申请中,大多数(71%)药品为新化学实体,29% 是治疗性生物药。申请的治疗领域中,肿瘤药占主导地位(25%)。EMA 和 FDA 是否在首次提交和审评时批准产品上市销售的决定对于 92%(98/107)的申请保持一致,对 8%(9/107)的申请不一致。关于 8% 的不一致决定,其中有 8 个产品仅获得 EMA 批准,1 个产品仅获得 FDA 批准。
研究还发现,有 15 件申请最初未获得一家或两家机构的批准:1 件申请未获得 EMA 批准,12 件未获得 FDA 批准,有 2 件申请双方都没有批准。在初步评估后,8 件申请被重新提交到 FDA,3 件被重新提交 EMA 药品评价委员会重新审查。对于重新审评的申请,FDA 批准了 8 件中的 7 件,EMA 批准了 3 件中的 2 件。总体而言,大多数第二次提交的申请最终都在两个机构获得批准。研究还指出,在审查相同的申请时,FDA 和 EMA 提出的基本科学和数据解释问题都有“显著相似性”。